 Previous posts have described the very complex pathways that create membranes to surround the entire cell and to build vital cell compartments and well known organelles. A post noted how different types of fatty membranes are built for each organelle with many different complex shapes. These include mitochondria, endoplasmic reticulum, the nucleus and many kinds of vesicles. Vesicles are used for transport, to combat invading microbes and to send information such as with neurotransmitters in the neuron.
Previous posts have described the very complex pathways that create membranes to surround the entire cell and to build vital cell compartments and well known organelles. A post noted how different types of fatty membranes are built for each organelle with many different complex shapes. These include mitochondria, endoplasmic reticulum, the nucleus and many kinds of vesicles. Vesicles are used for transport, to combat invading microbes and to send information such as with neurotransmitters in the neuron.
Possibly the most vital and complex vesicle is the lysosome. While originally thought to be a disposal and recycling factory for debris and misfolded material, it is in fact now known to be extremely vital for many critical cellular functions. Containing more than fifty complex enzymes it can break down any molecule and is vital for cellular signaling and response to infection.
Lysosomes are not just a sack of digestive enzymes, but rather a signaling hub related to all of the materials used in the cell. This signaling goes throughout the cell and the process where molecules that are produced are also tagged and transported to their appropriate cellular compartments. The lysosome is a sensory hub to determine what molecules are available, what has too much and what is needed. This includes all the molecules in metabolism of the cell such as amino acids to make proteins. A previous post noted that the molecule mTOR is one of the central hubs of regulating available materials, by measuring the amount of amino acids and regulating and stimulating the production of ribosomes to make more proteins. As will seen lysosomes and mTOR work together for many vital cellular projects.
Lysosomes and mTOR Regulate Each Other
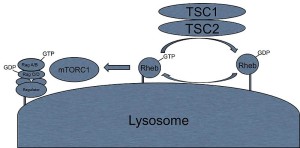
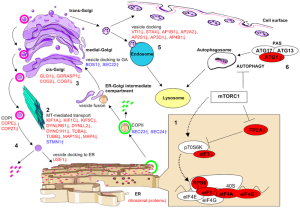
A previous post noted the vital importance of the regulatory molecule mTOR. mTOR a kinase enzyme—a protein enzyme that transfers phosphate energy particles like ATP—able to measure the amounts of amino acids and other molecules. The complex formed with mTOR (mTORC1) also regulates the amount of protein production. mTORC1 is active when there is need to stimulate more amino acids. But, when at rest it sits on the surface of the lysosome, which is also part of this regulatory system.
Other research showed that lysosomes are very responsive to changing environmental factors. In fact, recently, hundreds of genes were found for this complex lysosomal regulation. This network of genes is now called coordinated lysosomal expression and network or CLEAR. One critical protein was discovered that triggers the network as a transcription factor (TFEB or transcription factor EB). Then it was found that TFEB and TORC1 interact on the surface of the lysosome. mTORC1 suppresses the factor when it is not needed. But, once mTOR stops the suppression, the factor goes from the lysosome surface into the nucleus where it triggers the CLEAR network. These two powerhouses—mTOR and lysosomes—appear to regulate each other in a loop.
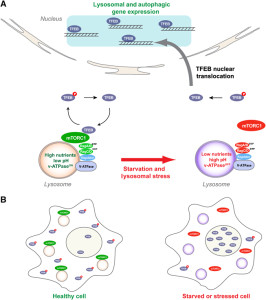 Current research is attempting to find out if lysosomes can be stimulated to clear debris more rapidly. This has implications for diseases where mis folded proteins accumulate, such as tau (see posts on tau diseases). One attempt is to boost TFEB or its gene network, which can function for long periods of time. Research is ongoing into the pathway that TFEB uses. A recent finding is that TFEB can increase the clearance of debris with lysosomes making sacs that take debris outside of the cell. This could help treatment of Parkinson’s, infections, cancer and obesity. One difficulty in affecting lysosomes in the brain includes treatments that can cross the blood brain barrier.
Current research is attempting to find out if lysosomes can be stimulated to clear debris more rapidly. This has implications for diseases where mis folded proteins accumulate, such as tau (see posts on tau diseases). One attempt is to boost TFEB or its gene network, which can function for long periods of time. Research is ongoing into the pathway that TFEB uses. A recent finding is that TFEB can increase the clearance of debris with lysosomes making sacs that take debris outside of the cell. This could help treatment of Parkinson’s, infections, cancer and obesity. One difficulty in affecting lysosomes in the brain includes treatments that can cross the blood brain barrier.
Lysosomes Vital for All Cellular Molecular Production
 Lysosomes are large complex sacs surrounded by membranes that have a large number of different enzymes to break down and recycle all kinds of molecules. They work on proteins, nucleic acids, carbohydrates and fats. They were thought to be both the waste disposal and the digestive department for all molecules. Molecules that are brought into the cell through membrane engulfment (endocytosis) are brought to the lysosomes. Debris and unnecessary molecules inside the cell are also tagged and brought to the lysosomes.
Lysosomes are large complex sacs surrounded by membranes that have a large number of different enzymes to break down and recycle all kinds of molecules. They work on proteins, nucleic acids, carbohydrates and fats. They were thought to be both the waste disposal and the digestive department for all molecules. Molecules that are brought into the cell through membrane engulfment (endocytosis) are brought to the lysosomes. Debris and unnecessary molecules inside the cell are also tagged and brought to the lysosomes.
Lysosomes are much more complex than previously thought. They are integrally involved in many vital cell processes. They are part of the secretory pathway (see the post on the production of membranes in cells) where they are vital in repairing membranes for the entire cell. They are a center of signaling throughout the cell, particularly related to the need for specific molecules. In this regard they work with mTOR to signal for the necessary amount of amino acids and other molecules. They are involved in the cell’s ability to secrete signals in vesicles outside of the cell. They are also involved in energy metabolism such as arranging the proper amount of appropriate molecules.
Many Lysosome Shapes
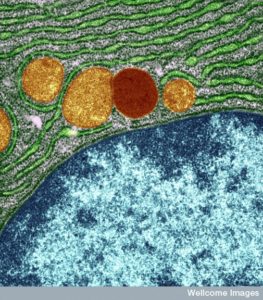 Lysosomes used to be considered round, but in fact, these large sacs can be in many different shapes. They can be very large and small depending on what job they are working on and what types of molecules they are recycling. The difference in size between the small and large sacs can be 10 times. In fact, they are a varied types of large sacs that are similar in that they serve the same type of function for digestion and recycling of particular material. This is another process that appears to have global direction without an obvious source.
Lysosomes used to be considered round, but in fact, these large sacs can be in many different shapes. They can be very large and small depending on what job they are working on and what types of molecules they are recycling. The difference in size between the small and large sacs can be 10 times. In fact, they are a varied types of large sacs that are similar in that they serve the same type of function for digestion and recycling of particular material. This is another process that appears to have global direction without an obvious source.
These organelles are in almost every animal cell. The lysosome maintains a particular pH (4.5 to 5.0) as an optimal amount to hydrolyze various molecules. This is similar to the way the stomach maintains a particular acid balance. But, the lysosome maintains in a singular vesicle. It contains a very large number of particular enzymes to break down almost every type of molecule that is found. It breaks down all large molecules (polymers).
Material Comes from Outside with Endocytosis and Inside with Autophagy
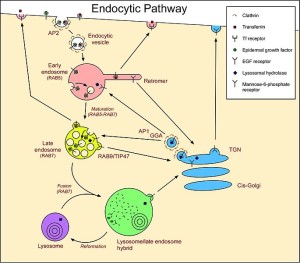
Lysosomes were first discovered by Christian de Duve (who won the Nobel Prize for this in 1974) as a waste removal of material from inside and outside the cell. Outside material comes in through sacs (endocytosis) and material inside uses the autophagy system (“self eating” system that will be discussed in more detail in the next post).
The endocytic process is in itself very complex and utilizes many different sizes and types of vesicles. It involves engulfing material to be brought into the cell for a variety of reasons, one being destruction of the material. Microbes often subvert this process to enter the cell and then avoid being brought to the lysosome for destruction. Some live out their lives in one of the vesicles (see post).
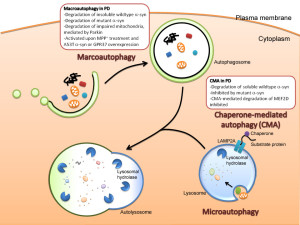 Autophagy is vital process whereby small specific sacs are created to eliminate specific materials. There are in fact multiple different ways that autophagy operates. It can occur as part of the lysosome itself. In this role small invaginations in the lysosome capture the relevant tagged particles to be destroyed or recycled. Another way it operates is along with chaperone molecules. These proteins work in the endoplasmic reticulum and fold proteins.
Autophagy is vital process whereby small specific sacs are created to eliminate specific materials. There are in fact multiple different ways that autophagy operates. It can occur as part of the lysosome itself. In this role small invaginations in the lysosome capture the relevant tagged particles to be destroyed or recycled. Another way it operates is along with chaperone molecules. These proteins work in the endoplasmic reticulum and fold proteins.
The major mechanism of autophagy is to create vesicles (with the strange name autophagosomes). This sac captures the tagged material and then eventually feeds into the lysosome by fusion where the materials are destroyed or recycled. Autophagy is highly involved in fighting cancer and stimulating particular immune responses. This will be described in the next post. It is difficult to determine where the regulation for this complex process resides since it utilizes many different vesicles.
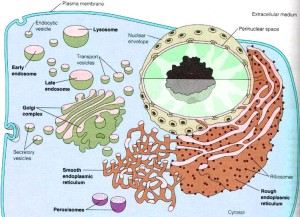 The large amount of different proteins enzymes is manufactured in the endoplasmic reticulum which, along with the Golgi, is part of the secretory system that produces all different kinds of fatty membranes. These proteins are sent to the Golgi were they are tagged with mannose 6-phosphate, and placed in small transportation vesicles. As these smaller vesicles fuse together they become the larger lysosomes which become acidic. This particular tag helps create the acidic environment in the lysosome.
The large amount of different proteins enzymes is manufactured in the endoplasmic reticulum which, along with the Golgi, is part of the secretory system that produces all different kinds of fatty membranes. These proteins are sent to the Golgi were they are tagged with mannose 6-phosphate, and placed in small transportation vesicles. As these smaller vesicles fuse together they become the larger lysosomes which become acidic. This particular tag helps create the acidic environment in the lysosome.
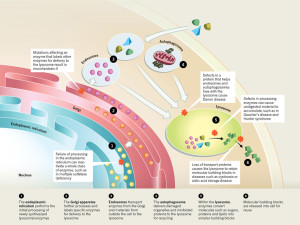
The nuclear genetic networks that produce this large number of different enzymes can have mutations, which create at least thirty different diseases called lysosomal storage diseases (LSDs). With faulty enzymes, then specific material is not able to be fragmented and material accumulates that shouldn’t. This unwanted material causes problems in particular cells that become cancer, and heart and brain diseases.
Many diseases
Many different enzymes in lysosomes can have mutations that affect one of the many pathways whereby the lysosome degrades a particular molecule. There are now at least 50 different lysosome storage diseases (LSDs) that have many different effects including abnormal calcium levels, increasing destructive oxygen reactive molecules, disturbance of the lipid pathways that make membranes, and various types of inflammation. Some of these are from one gene mutation. These create unique diseases.
But other diseases arise from interactions with other organelles:
- One is when the abnormal protein is the one that places labels on molecules in order to deliver it to the lysosome (mucolipidosis-II)
- There are diseases caused by the proteins that allow autophagy vesicles to fuse with the lysosome.
- When enzymes in the lysosome don’t work to break down molecules, then abnormal molecules accumulate. (Gaucher’s disease and Hunter syndrome)
- Abnormalities in proteins that transport molecules hurts lysosomes (cystinosis and sialic acid storage disease)
- Failure in the endoplasmic reticulum stops function of many enzymes at once (Multiple sulfatase deficiency)
Lysosomes Vital in Brain Disease
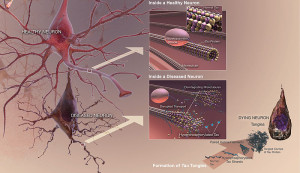 One well known lysosome disease is Niemann-Pick a rare lysosomal storage disease (LSD). It causes a degenerative brain disease that is a form of Alzheimer’s dementia in children. In fact, children with this disease have the same tangles of the misfolded version of tau. Please see other posts for the description of tau—a molecule that stabilizes microtubules in neurons—and its abnormal versions that cause various dementias.
One well known lysosome disease is Niemann-Pick a rare lysosomal storage disease (LSD). It causes a degenerative brain disease that is a form of Alzheimer’s dementia in children. In fact, children with this disease have the same tangles of the misfolded version of tau. Please see other posts for the description of tau—a molecule that stabilizes microtubules in neurons—and its abnormal versions that cause various dementias.
Brain cells are specially affected by lysosome alterations because neurons don’t divide and therefore recycling problems build up over the lifetime of the neuron. In the body, cells that accumulate too much can be replaced.
Another brain disease associated with lysosomes is fronto temporal dementia. This is related to a lipid pathway with a molecule called progranulin and is now found to be also on the lysosome, regulated by the same TFEB.
Early onset Alzheimer’s also has an abnormal protein presenilin on the lysosome membrane. This is needed to break down debris such as those that are part of Alzheimer’s. Recently, lysosome enzymes have been connected to six other brain diseases.
An altered enzyme (NPC1) causes cholesterol to accumulate in the lysosomes and therefore in many different cells throughout the body. A very strange coincidence connects the same gene to cholesterol and Ebola. This gene causes an alteration in a cholesterol enzyme in the lysosome. But at the same time it also affects Ebola viruses ability to enter human cells. Recently, this very strange situation was discovered because the same protein is a receptor on the lysosome inside the cell that allows Ebola to attach and enter the cell. Very recent research showed that NPC1 has direct interactions with the surface coat of the virus.
Parkinson’s and Gaucher’s Diseases
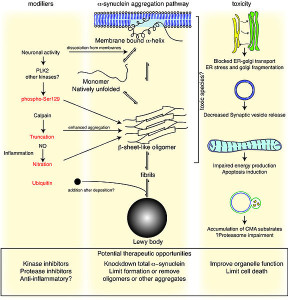
One of the best known lysosomal storage disease is called Gaucher’s. In this disease a lysosome enzyme in the lipid pathway is altered. Recently, this has been associated with Parkinson’s disease, the second most common brain degenerative disease (1% of the population). In Parkinson’s there is accumulation of an abnormal protein called a-synuclein. The varied causes of Parkinson’s are now known but in 20% several genes appear to be related to familial types.
In Gaucher’s disease, abnormal genes alter the lipid glucosylceramide. There are three types. The most common affects liver and spleen and a less common types affect the brain leading to early death. Recently, it has been found that the same gene is a risk factor for Parkinson’s as well as Gaucher’s even though the diseases do not appear with the same symptoms.
Since it is not clear how this one gene can affect both diseases, there are many different hypotheses and lines of research.
 The abnormal gene makes a version of the lipid enzyme that isn’t properly folded. It doesn’t get rid of the lipid it is supposed to and also interacts with a different pathway that it normally doesn’t. Protein folding problems have been associated with most other degenerative brain diseases—Alzheimer’s, Mad Cow, and Parkinson’s. In Gaucher’s, the abnormal kinase accumulates in the endoplasmic reticulum. Animal models of Parkinson’s have the same accumulation of the lipid kinase with the a-synuclein. The increased abnormal glucocerebrosidase enzyme could increase a-synuclein clumps or interfere with its removal. Some mouse studies show that without the kinase there are still symptoms of Gaucher’s and sometimes Parkinson’s. Both diseases can occur without the mis folded proteins which implies that the lysosome is even more critical.
The abnormal gene makes a version of the lipid enzyme that isn’t properly folded. It doesn’t get rid of the lipid it is supposed to and also interacts with a different pathway that it normally doesn’t. Protein folding problems have been associated with most other degenerative brain diseases—Alzheimer’s, Mad Cow, and Parkinson’s. In Gaucher’s, the abnormal kinase accumulates in the endoplasmic reticulum. Animal models of Parkinson’s have the same accumulation of the lipid kinase with the a-synuclein. The increased abnormal glucocerebrosidase enzyme could increase a-synuclein clumps or interfere with its removal. Some mouse studies show that without the kinase there are still symptoms of Gaucher’s and sometimes Parkinson’s. Both diseases can occur without the mis folded proteins which implies that the lysosome is even more critical.
Another finding is that a different lysosomal kinase related to Niemann Pick (SMPD1) can increase the amount of Parkinson’s.
These studies together point in the direction of general lysosome function being involved in the cause of Parkinson’s. One of the most important functions of lysosomes involves the process called autophagy. This is the vital connection of collecting debris in the cell and then bringing it to the lysosome for clearance. The complex interactions of autophagy with metabolism and cancer will be discussed in the next post.
A current research project is testing the two possibilities. Does mis folded glucocerebrosidaces cause accumulation of a-synuclein as a cause of Parkinson’s. Or does abnormal glucocerebrosidase cause a global change in the way the lysosome operates that causes Parkinson’s. It is also possible that both mechanisms are involved.
A future post will discuss the complex ways that autophagy is related to cancer, inflammation, and brain disease.
Intelligent Lysosomes Are More than Waste Disposal
 It is difficult to imaging where the direction comes for the vast amount of functions of the lysosomes. But, this also can be said of the entire secretory system that the lysosome is part of. This includes endoplasmic reticulum producing proteins with ribosomes, as well as many kinds of lipids for membranes. It includes transporting and tagging these molecules and membranes in the Gogli apparatus and then transporting them all over the cell.
It is difficult to imaging where the direction comes for the vast amount of functions of the lysosomes. But, this also can be said of the entire secretory system that the lysosome is part of. This includes endoplasmic reticulum producing proteins with ribosomes, as well as many kinds of lipids for membranes. It includes transporting and tagging these molecules and membranes in the Gogli apparatus and then transporting them all over the cell.
But, direction from somewhere also connects mTOR and the lysosome to regulate all of the needed materials throughout the cell. As will be seen the same kind of distributed regulation must occur with the autophagy process that can operate in the lysosome by capturing material at the membrane, but also separately in its own vesicles (autophagosome). There is an underlying direction across the entire secretory pathway including ER, Golgi, mitochondria, lysosomes and all the other compartments with membranes.