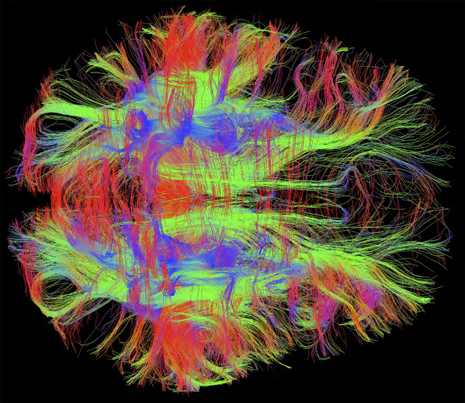
 Mis folded proteins transmitted in circuits throughout the brain might explain many degenerative brain diseases. Considerable evidence now points to the fact that critical mis folded proteins, once they appear, can act like prions by attracting other similar proteins and stimulating them to alter their structure as well. These abnormal mis folded proteins either individually, or in combinations of two or more, can form fibers and clumps, which are the hallmarks of degenerative brain disease. What is striking about the new research is that the toxic molecules or clumps are then transmitted along axons and across synapses. They end up in entire large synaptic circuits that are the known regions for degenerative brain diseases such as Alzheimer’s, Parkinson’s, ALS, Fronto-temporal Dementia, brain trauma encephalopathy and other forms of dementia.
Mis folded proteins transmitted in circuits throughout the brain might explain many degenerative brain diseases. Considerable evidence now points to the fact that critical mis folded proteins, once they appear, can act like prions by attracting other similar proteins and stimulating them to alter their structure as well. These abnormal mis folded proteins either individually, or in combinations of two or more, can form fibers and clumps, which are the hallmarks of degenerative brain disease. What is striking about the new research is that the toxic molecules or clumps are then transmitted along axons and across synapses. They end up in entire large synaptic circuits that are the known regions for degenerative brain diseases such as Alzheimer’s, Parkinson’s, ALS, Fronto-temporal Dementia, brain trauma encephalopathy and other forms of dementia.
Previous posts have discussed the prion—a protein that folds abnormally and becomes toxic. Prions attract and alter other normal proteins and form an expanding crystal of abnormal proteins. Neurons appear to secrete and reabsorb prions sending them throughout the brain. Now, it is shown that other mis folded proteins, while not exactly prions, behave like prions and produce brain disease.
Cleaning The Brain of Mis Folded Proteins
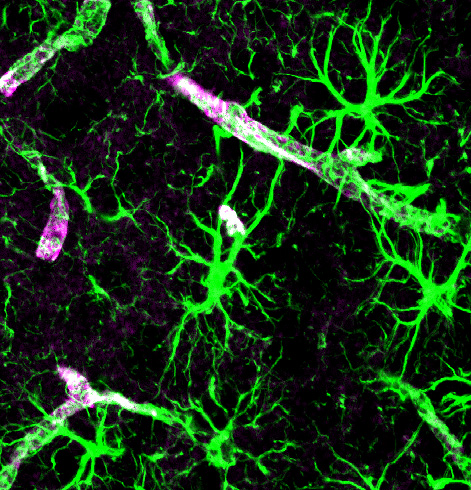 As abnormal proteins appear, usually they are cleaned by two major mechanisms. Both cleaning mechanisms help avoid dementia by getting rid of the particles that form toxic molecular clumps in or near the damaged neurons.
As abnormal proteins appear, usually they are cleaned by two major mechanisms. Both cleaning mechanisms help avoid dementia by getting rid of the particles that form toxic molecular clumps in or near the damaged neurons.
The first method consists of microglia and other immune cells gobbling the debris.
The second is the newly discovered brain cleaning mechanism called the “glia lymphatic” or “glymphatic system.” It consists of a convective flow of the salty fluid between the brain cells from arteries and cerebral spinal fluid to veins. A very surprising discovery is that during sleep neurons shrink to allow the extra cellular fluid space to increase by 60%, which greatly increases the cleaning.
Mis Folded Proteins that Produce Clumps and Brain Disease
Tau
 The protein tau usually holds together microtubules providing scaffolding and transport of material along the axon. An alteration of tau occurs when a phosphate is added to one of its amino acids changing its shape and its properties. It fact, there are many different forms of toxic tau. In Alzheimer’s, phosphorylated tau becomes insoluble and forms neurofibrillary tangles. Another well-known tauopathy is Frontotemporal dementia, FTLD-tau. At least six different forms of abnormal tau have been found, made up of patterns of three repeats and four repeats that occur in different brain diseases. Alzheimer’s has 3R and 4R, while Picks disease has only 3R.
The protein tau usually holds together microtubules providing scaffolding and transport of material along the axon. An alteration of tau occurs when a phosphate is added to one of its amino acids changing its shape and its properties. It fact, there are many different forms of toxic tau. In Alzheimer’s, phosphorylated tau becomes insoluble and forms neurofibrillary tangles. Another well-known tauopathy is Frontotemporal dementia, FTLD-tau. At least six different forms of abnormal tau have been found, made up of patterns of three repeats and four repeats that occur in different brain diseases. Alzheimer’s has 3R and 4R, while Picks disease has only 3R.
The toxicity can occur either by several tau molecules or fibrils made from many tau molecules. Recently, it has been discovered that tau can be released from neurons and taken up by the next neuron.
Amyloid-β
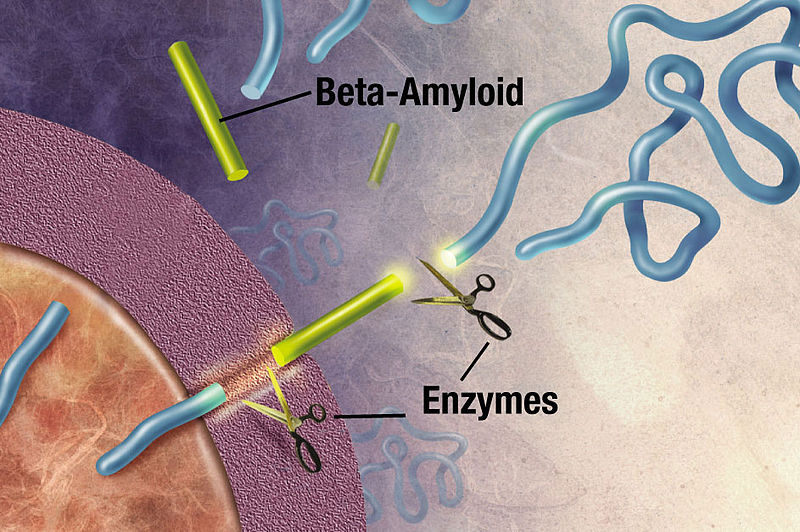 Amyloid-β is important for the synapse, but its exact role is unknown. Amyloid beta is a peptide (small protein) of approximately 40 amino acids. Normally, amyloid is not toxic and is involved in protection against oxidation, regulation of cholesterol transport, and serves as a transcription factor. It is cleared by the glymphatic system. APP is a large protein that sits in the membrane of the neuron near the synapse and is cut by the enzyme gamma secretase producing amyloid-β particles of various sizes. A specific mutation of this enzyme produces 42-sized amyloid-β that seems to form the toxic clumps.
Amyloid-β is important for the synapse, but its exact role is unknown. Amyloid beta is a peptide (small protein) of approximately 40 amino acids. Normally, amyloid is not toxic and is involved in protection against oxidation, regulation of cholesterol transport, and serves as a transcription factor. It is cleared by the glymphatic system. APP is a large protein that sits in the membrane of the neuron near the synapse and is cut by the enzyme gamma secretase producing amyloid-β particles of various sizes. A specific mutation of this enzyme produces 42-sized amyloid-β that seems to form the toxic clumps.
α-synuclein
 Another protein α-synuclein accumulates in Lewy bodies as a hallmark of Parkinson’s and a special form of dementia called Lewy body dementia. α-synuclein is a molecule in the pre synaptic neuron that is critical for forming the sacs (vesicles) that carry neurotransmitters. It can be secreted from neuronal cells in sacs. It is, also, released by dying neurons.
Another protein α-synuclein accumulates in Lewy bodies as a hallmark of Parkinson’s and a special form of dementia called Lewy body dementia. α-synuclein is a molecule in the pre synaptic neuron that is critical for forming the sacs (vesicles) that carry neurotransmitters. It can be secreted from neuronal cells in sacs. It is, also, released by dying neurons.
α-synuclein can also be transmitted out of neurons in vesicles, enter another cell and then, when inside the cell, break out of the vesicle disrupting the mitochondria.
SOD1
ALS (amyotrophic lateral sclerosis of Lou Gehrig’s Disease) comes in two basic varieties. The 10% that runs in families has mutations of superoxide dismutase-1 (SOD1). The clumps in this case are from SOD1, which arises in microglia and astrocytes. These clumps, while secreted from microglia, also, activate microglia and stimulate neuronal death. Microglia that produce SOD1 are impaired and cannot clean up debris. Studies now show that general infections increase the ALS process.
TDP43
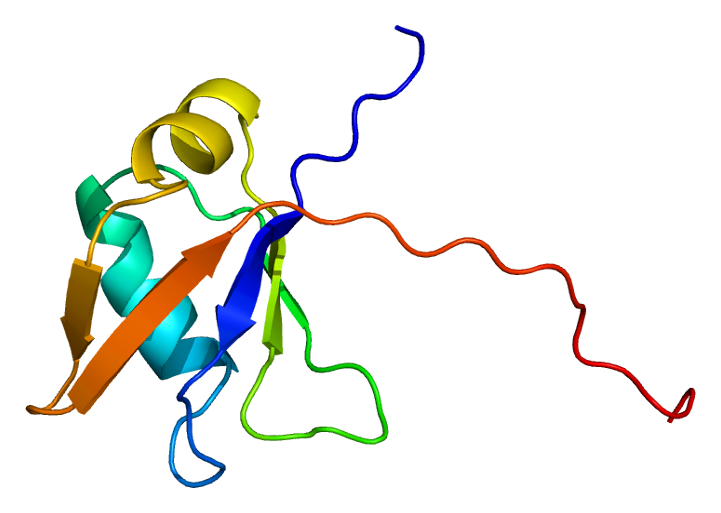 Another protein, TDP43 (TAR DNA-binding protein 43), is the cause of many ALS sporadic cases. It accumulates in the destroyed neurons. The normal version of this molecule binds DNA as a repressor and, also, binds RNA. It has multiple functions in alternative splicing of messenger RNA and regulation of protein production. The abnormal version has multiple extra phosphates, as well as attached ubiquitin molecules. It exists in many diseases along with tau, but especially in Fronto-temporal dementia (FTLD-TDP) and ALS.
Another protein, TDP43 (TAR DNA-binding protein 43), is the cause of many ALS sporadic cases. It accumulates in the destroyed neurons. The normal version of this molecule binds DNA as a repressor and, also, binds RNA. It has multiple functions in alternative splicing of messenger RNA and regulation of protein production. The abnormal version has multiple extra phosphates, as well as attached ubiquitin molecules. It exists in many diseases along with tau, but especially in Fronto-temporal dementia (FTLD-TDP) and ALS.
Polyglutamine Repeats
An altered huntingtin protein with polyglutamine repeats is the hallmark of Huntington’s disease.
Similar Properties of Mis Folded Proteins
All of these different proteins are altered from a normal state into molecules that form clumps. Small numbers of the proteins combine to form fibers or large macromolecules that are not soluble and not successfully cleared by the two cleaning mechanisms. All act as prions in that they affect the other normal proteins to fold in abnormal ways increasing clumps. The process that alters normal proteins spreads and is then transmitted to other neurons and glia cells.
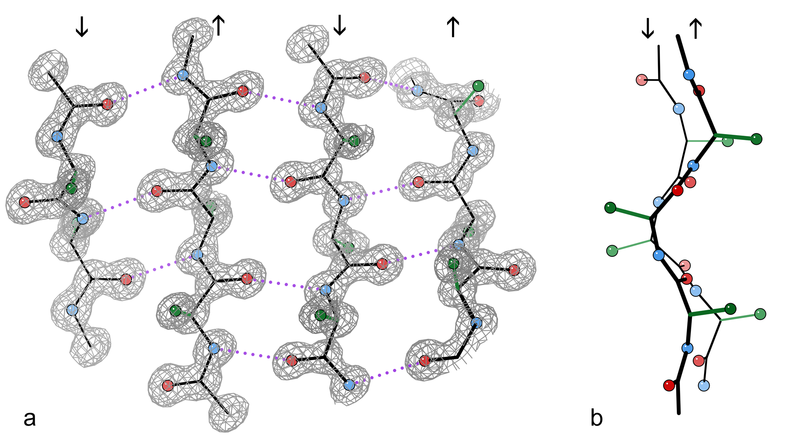
All clumps are made of protein filaments that don’t branch called β strands. They then become twisted and form β sheets. These are termed amyloids. The TDP43 strands are often straight, but can become twisted amyloids as well. One section of the molecule functions as a prion. Recent research shows that some of the smaller molecules, before they become fibers and clumps, are the most dangerous. This appears to be the case in Alzheimer’s where the small oligomer of a certain size is the most dangerous type of amyloid-β.

Proteins fold in four levels (see post on Protein Folding for details). The hydrophobic forces bring about secondary folding which most often takes the form of alpha and beta sheets. Beta-strands are a normal structural unit of protein beta sheets. This is an extended stretch of polypeptide chain typically 3 to 10 amino acids long that forms hydrogen bonds with other beta-strands in the same beta-sheet. Beta sheets consist of beta strands connected laterally by at least two or three backbone hydrogen bonds. Beta sheets are formed from multiple attached amyloid-β molecules.
Once the secondary structure occurs, then other forces from the unique sequence of amino acid side chains formthe tertiary and quaternary folds. Folding is so complex that chaperone molecules are needed and the most advanced computers cannot calculate how the folding will occur from the amino acid sequence. (see Post of Protein Folding)
Prions
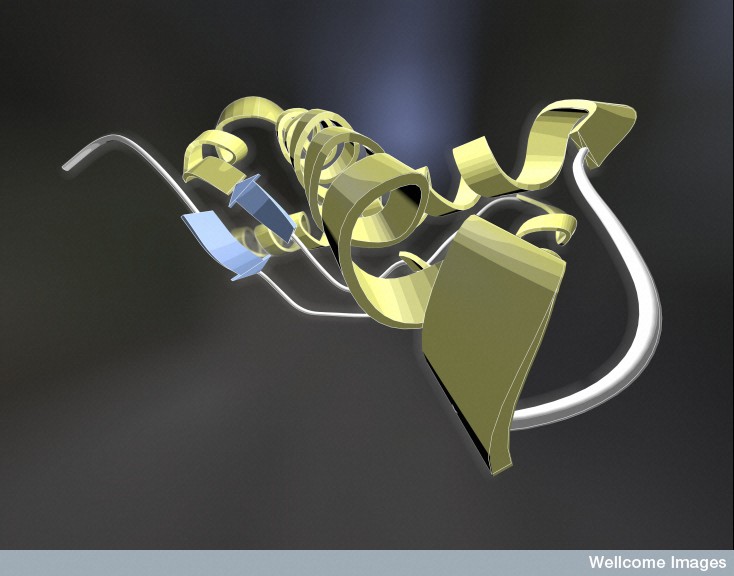 Prions come from the normal protein molecule PrPc, which is altered into the abnormal prion PrPSc. Prions attract and then interact with other molecules of the same type forming an enlarging crystal-like clump. The normal protein that becomes a prion has an alpha helix. The prion version of the same protein has beta sheets, a completely different structure.
Prions come from the normal protein molecule PrPc, which is altered into the abnormal prion PrPSc. Prions attract and then interact with other molecules of the same type forming an enlarging crystal-like clump. The normal protein that becomes a prion has an alpha helix. The prion version of the same protein has beta sheets, a completely different structure.
The spreading prions produce human Mad Cow disease (Creutzfeldt-Jakob Disease) and related brain diseases in sheep, goats, cattle and deer (see post on Prions). Different versions in various animals produce unique brain diseases. These proteins are infectious, transferring the active prion from animal to animal by ingestion. The human form of Creutzfeldt-Jakob killed more 200 people from eating infected cows’ meat. There are, also, occasional “sporadic” cases. Among 7000 people who were injected with growth hormone taken from deceased human pituitaries, 24 developed the Creutzfeldt-Jakob prion disease.
PrPc has many different normal important physiological roles. It is involved in maintaining myelin formation; developing T cells and is critical in stem cells that make blood cells and M cells of the intestine immune centers. PrPc is involved in virus reproduction and modulation of inflammation in the CNS. It can modulate the immune response to its own advantage and can inhibit the function of macrophages, which engulf  or phagocytize other debris.
or phagocytize other debris.
If eaten, prions reproduce and grow in the intestinal lymph tissues (Peyer’s patches) then tonsils and spleen. Peripheral lymph tissues are innervated by sympathetic nerves and most of the time prions spread into the brain through the autonomic nervous system. Prions enter nerves that are closest to the lymph tissues and spread from there. Once in the brain, prions spread along neuronal circuits.
Prion-Like Proteins, Prionoids and Non Prion Proteins
 This same method of transmission occurs in other mis folded proteins. These are sometimes called prion-like particles. It is now known that neurons expel and pick up vesicles with prion-like proteins. A receptor has been found related to tau and amyloid-β for this purpose.
This same method of transmission occurs in other mis folded proteins. These are sometimes called prion-like particles. It is now known that neurons expel and pick up vesicles with prion-like proteins. A receptor has been found related to tau and amyloid-β for this purpose.
Neurons secrete prion-like proteins in vesicles (like neurotransmitters) and other neurons pick up these vesicles. Very recently, it was observed that prion-like proteins, such as amyloid-β, attach and clump on neuronal receptors consisting of the original prions and material from the extra cellular matrix (see post on Extra Cellular Matrix.) It was, also, shown that prion-like proteins break open vesicles inside neurons, escape, and attack mitochondria.
The new prion-like particles are sometimes called prionoids, which are transmitted from cells to other cells in the brain. While they have the same type of transmission throughout the brain circuits, there is little evidence of infections from one person to another. Among the 7000 patients injected with human pituitary that produced prion disease, there were no cases of tau or α-synuclein, which implies that, as far as we know, these are not transmitted between people. They are transmitted between neurons in one brain. As well, there has been no animal transmitted disease similar to Mad Cow with these mis folded proteins.
However, when fetal neuron stem cells were transplanted into Parkinson’s brains, these new neurons, also, developed the same Parkinson’s pathology with Lewy bodies and brain damage.
Origination of the Mis Folded Protein
 Thus far, we do not know how mis-folded proteins originate. But, once they occur, they become a source producing more of the same mis folded proteins. This is called template directed production of the abnormal proteins. They attract normal molecules and then alter them in the template.
Thus far, we do not know how mis-folded proteins originate. But, once they occur, they become a source producing more of the same mis folded proteins. This is called template directed production of the abnormal proteins. They attract normal molecules and then alter them in the template.
Abnormal tau was injected into mice brains and they seeded a production of more abnormal tau. In one experiment, synthetic abnormal tau fibers caused more disease. What has been surprising in the new research is the many sub varieties of tau, α-synuclein and Amyloid-β that produce different brain effects. They produce different types of seeding and different types of brain damage.
Transmission of Mis Folded Protein
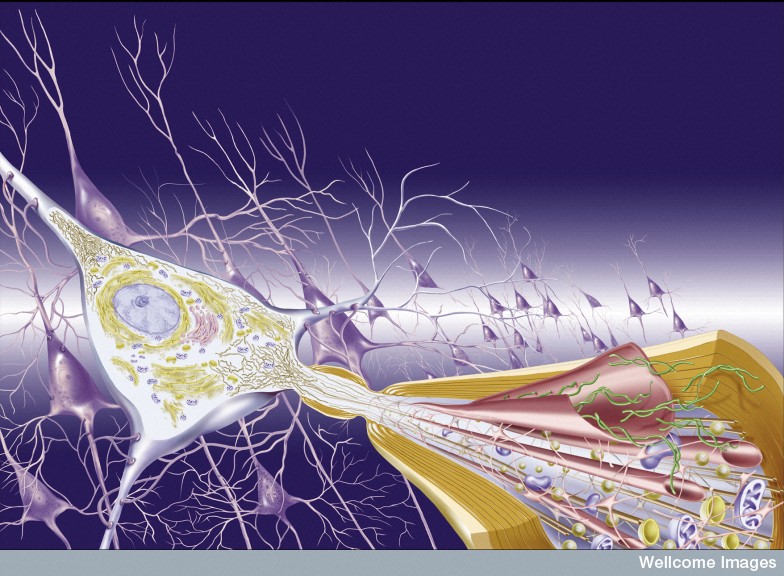 First, it was noted that prions introduced through surgery could spread from the eye along visual pathways. Spread was found to occur along the vagus nerve to the medulla, pons and midbrain. In transplants to treat Parksinson’s, the new neurons develop the same disease from the local cells. Injection of abnormal tau, α-synuclein and amyloid-β in animal experiments shows spread into the new brain.
First, it was noted that prions introduced through surgery could spread from the eye along visual pathways. Spread was found to occur along the vagus nerve to the medulla, pons and midbrain. In transplants to treat Parksinson’s, the new neurons develop the same disease from the local cells. Injection of abnormal tau, α-synuclein and amyloid-β in animal experiments shows spread into the new brain.
The transmission paths of various mis folded proteins are very complex and not fully understood. For example, there are many interactions between the effects of tau, α-synuclein and amyloid-β. Extracellular molecules, called HSPG, bind tau and bring it into the cell. HSPGs are, also, essential for cell-to-cell spread of α-synuclein. HSPG is part of amyloid plaques and neurofibrillary tangles. HSPGs appear to increase the formation of amyloid-β by inhibiting the enzyme cutting APP. It, also, interacts with apolipoprotein E (APOE), which is known as a major risk factor for Alzheimer’s.
Amyloid-β, which exists outside of the neuron in the extracellular space, has been shown to travel to distant brain regions. Other diseases, also, show this spread but begin as clumps inside a neuron or glial cell and must be released from the cell. It is possible that larger clumps of tau fibrils are released from the cell. But, now research shows that smaller pieces of tau, even one molecule, can be sent from the cell via vesicles, called exosomes. This has been shown for both α-synuclein and tau. In fact, there has been evidence of the exosomes being controlled by receptors for this purpose.
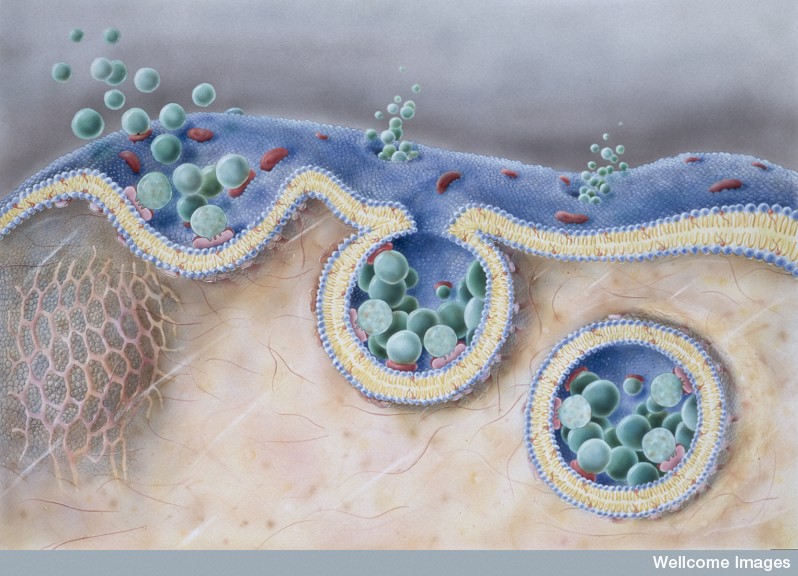 Endocytosis has been observed in the uptake of tau, SOD1 monomers (just one molecule) and α-synuclein single, double and triple molecules (oligomers) and fibrils (many molecules). Altered huntingtin protein seems to pass through the membrane without needing vesicles. Therefore, it appears that the transfer can occur with one molecule, small clumps (oligomers) or larger clumps of fibrils.
Endocytosis has been observed in the uptake of tau, SOD1 monomers (just one molecule) and α-synuclein single, double and triple molecules (oligomers) and fibrils (many molecules). Altered huntingtin protein seems to pass through the membrane without needing vesicles. Therefore, it appears that the transfer can occur with one molecule, small clumps (oligomers) or larger clumps of fibrils.
There is now clear evidence that tau and α-synuclein spread from neuron to neuron through synaptic connections. What is interesting is that in studies where fibrils have been injected into different brain regions, they all transmit the disease. It does not seem to be a characteristic of the specific neuron type or region, but rather the disease starts the spread from the point where it originates. Injection of α-synuclein or tau individual molecules or fibrils does not cause clumps where the injection took place, but only after being transmitted to distant sites.
The circuits connected with the origination site become the direction of the transmission of the disease. Therefore, neurons near the injection site, but not part of a circuit, would not develop the disease, but far away regions would, if they are connected. This spread can start outside the brain and travel by neuronal circuits to the brain. α-synuclein ingested in the intestine can spread from enteric neurons and the vagus to the brain causing Parkinson’s. Studies have not determined that the spread occurs through glia, or the blood brain barrier, or fluids. Current research is consistent with the spread by neuronal circuits.
Transmission along the neuron’s axon can be in either direction often by the special microtubule dynein motors. (see post Complexity of Axon Transport).
Mis Folded Proteins Circuits in Human Brain Disease
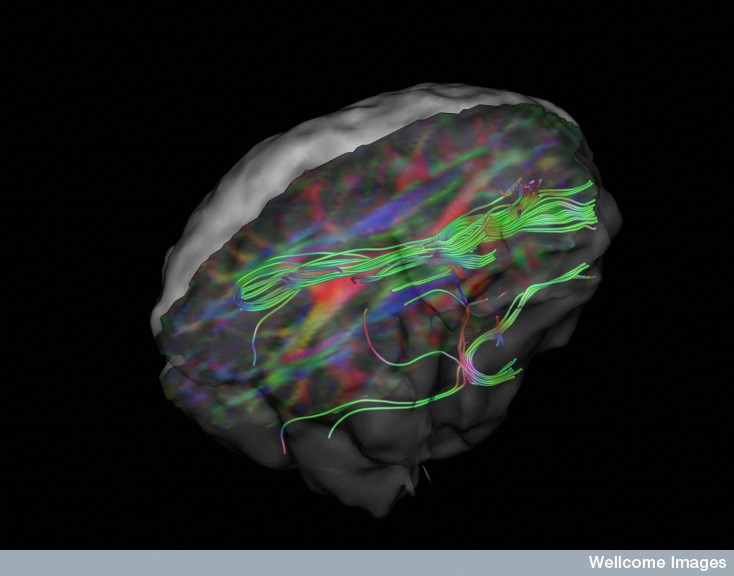 Imaging studies show that clumps appear progressively in specific different brain regions. Over time the clumps grow. In Alzheimer’s, the tau clumps start in specific regions like the locus coeruleus and entorhinal cortex then travel to other regions to the neo cortex. In the brain disease from multiple concussions (often from professional contact sports) a different abnormal tau can start in one area near the vessels of the cortex surface and then spread to many areas including other areas of the cortex, spinal cord, brainstem, and basal ganglia. The movement of various lesions are different.
Imaging studies show that clumps appear progressively in specific different brain regions. Over time the clumps grow. In Alzheimer’s, the tau clumps start in specific regions like the locus coeruleus and entorhinal cortex then travel to other regions to the neo cortex. In the brain disease from multiple concussions (often from professional contact sports) a different abnormal tau can start in one area near the vessels of the cortex surface and then spread to many areas including other areas of the cortex, spinal cord, brainstem, and basal ganglia. The movement of various lesions are different.
Amyloid-β travels in an opposite direction of tau in the brain. In Alzheimer’s, amyloid-β starts in the neocortex, travels through other regions and cortex and then to the brainstem and cerebellum. Also, Amyloid-β may be involved in producing tau and tau may then alter amyloid-β’s affects. But, it is not clear exactly how they interact.
α-synuclein with associated Lewy bodies, Parkinson’s disease and Lewy Body dementia start in the olfactory bulb and vagus in the medulla (ventral) and move to the pons, midbrain and neocortex . The hallmark of Parkinson’s disease is the destruction of the dopamine neurons of the substantia nigra in the midbrain and is late in onset.
Those who develop Parkinson’s can have α-synuclein start in the intestinal neurons (Meissner’s plexus) and below the mandible. Studies have been inconsistent exactly where the first sites are (abdomen, olfactory or vagus nerve). Some think that it starts in two separate places at the same time.
Unique Multiple Directions of TDP43 Spread
 In the sporadic form of ALS, the mis folded protein is TDP43, which progresses downward starting in the motor cortex to the spinal cord and brainstem. Later it spreads to other areas of the cortex. Surprisingly, TDP43 is, also, seen in Alzheimer’s along with tau and amyloid-β, but travelling in a totally different direction from the amygdala to the hippocampus, cortex and basal ganglia. An entirely different pattern of travel for this same TDP43 occurs in Fronto-temporal dementia starting in the frontal area and spreading backwards through the cortex to the occipital region. In traumatic encephalopathy (from boxing, football and multiple concussions) it travels throughout the cortex and spinal cord.
In the sporadic form of ALS, the mis folded protein is TDP43, which progresses downward starting in the motor cortex to the spinal cord and brainstem. Later it spreads to other areas of the cortex. Surprisingly, TDP43 is, also, seen in Alzheimer’s along with tau and amyloid-β, but travelling in a totally different direction from the amygdala to the hippocampus, cortex and basal ganglia. An entirely different pattern of travel for this same TDP43 occurs in Fronto-temporal dementia starting in the frontal area and spreading backwards through the cortex to the occipital region. In traumatic encephalopathy (from boxing, football and multiple concussions) it travels throughout the cortex and spinal cord.
Clumps Travel in Projection Neurons
 A previous post described the fantastic complexity of more than a thousand different neuron types. We don’t know how many different types of neurons exist. There are a wide variety of projection neurons, a very special type of neuron with extremely long axons that project from one part of the brain to entirely different regions. Projection neurons are excitatory and critical for all major sensory, cognitive and motor functions. In fact, projection neurons are determined during fetal development by very complex triggers that are just now being discovered.
A previous post described the fantastic complexity of more than a thousand different neuron types. We don’t know how many different types of neurons exist. There are a wide variety of projection neurons, a very special type of neuron with extremely long axons that project from one part of the brain to entirely different regions. Projection neurons are excitatory and critical for all major sensory, cognitive and motor functions. In fact, projection neurons are determined during fetal development by very complex triggers that are just now being discovered.
During fetal development, many complex signals stimulate the projection neuron’s axon to migrate very long distances. It is possible that this uniqueness provides the environment for the origination and transit of mis folded clumps in the major brain diseases.
 The neurons that develop clumps of tau, amyloid-β and TDP43 are very long and thin projection neurons with only a small amount of myelin. Projection neurons that are short and fat and have a lot of myelin don’t have clumps. This may be because long thin un-myelinated neurons require a very great amount of energy to support the axon transport and other ion channels. Another type of neuron that develops clumps has a very large long axon with a great amount of mitochondria, which, also, uses a lot of energy.
The neurons that develop clumps of tau, amyloid-β and TDP43 are very long and thin projection neurons with only a small amount of myelin. Projection neurons that are short and fat and have a lot of myelin don’t have clumps. This may be because long thin un-myelinated neurons require a very great amount of energy to support the axon transport and other ion channels. Another type of neuron that develops clumps has a very large long axon with a great amount of mitochondria, which, also, uses a lot of energy.
Brain regions with a large amount of connectivity are susceptible to the transmitted clumps. The spread could be from direct molecule-to-molecule contact, by contact at the synapses and by transport along the axon (see post on Complexity of axon transport).
Are Glia Involved in the Spread of Clumps
Other posts have described astroctyes, microglia and oligodendrocytes playing important roles in degenerative brain diseases. Microglia can either be fighting against the clumps, trying to surround and limit them, or in pathological situations enhancing them. Some studies show that astrocytes engage in endocytosis with α-synuclein from neurons altering immune function. This same transfer occurs from axons to oligodendrocytes. Tau was, also, transmitted to astrocytes and oligodendrocytes. amyloid-β was transmitted to astrocytes. But, glia’s role is not yet clear.
Transmission of Mis Folded Proteins in Brain Disease
 Transmission of toxic protein clumps is a new theory for degenerative brain disease. While it is a promising lead, the fact that the origination of the abnormal clumps is not clear still leaves the question open.
Transmission of toxic protein clumps is a new theory for degenerative brain disease. While it is a promising lead, the fact that the origination of the abnormal clumps is not clear still leaves the question open.
In this view, there are specific places where a template starts the mis folding. Once started, the mis folded proteins produce other mis folding. These single, double and triple sized molecules, fibrils, filaments and clumps are, then, transmitted along axons, synapses and neuronal connections. It is not clear what is transmitted—the smaller pieces, the filaments, the clumps, or all of them.
If this is the primary mechanism of many brain diseases, it will, hopefully, open up avenues of treatment at the origination site or along the transmission routes.