 Varicella zoster virus is an alpha herpes virus that has been described in a previous post as having remarkable capabilities and a very complex lifestyle—the ability to travel up and down axons in a neuron, to move in and out of several different types of cells, to travel and multiply in T cells, to fool several different nuclear pore complexes, to commandeer microtubule motors, and to alter its own behavior pattern in different settings. How can the remarkable intelligent varicella virus know how to do all of these complex tasks?
Varicella zoster virus is an alpha herpes virus that has been described in a previous post as having remarkable capabilities and a very complex lifestyle—the ability to travel up and down axons in a neuron, to move in and out of several different types of cells, to travel and multiply in T cells, to fool several different nuclear pore complexes, to commandeer microtubule motors, and to alter its own behavior pattern in different settings. How can the remarkable intelligent varicella virus know how to do all of these complex tasks?
Varicella is specific to humans and causes chickenpox and shingles. The complex molecular mechanisms for each behavior of the remarkable intelligent varicella virus are just now being discovered. How can such a simple organism, which is basically just a small piece of DNA with protein covers, take control of multiple large complex cells, such as human T cells, neurons and skin cells? How can varicella know how to use so many different techniques in sequence?
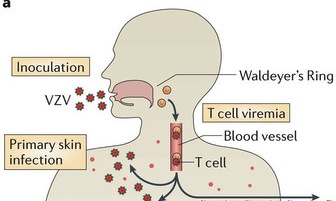 The infection begins in upper respiratory mucosa cells, spreads to the lymph and the tonsils, and, then, is carried by T cells in the blood to the skin where it causes a widespread skin rash. The virus is, also, able to enter sensory neurons, take over transport motors and travel to the ganglion cell bodies. It then completely changes behavior and becomes dormant in the neuron for years. When it awakens, varicella, again, travels on the axon microtubule transport system to the skin to cause the painful zoster rash in the region that neurons innervate. The virus is plentiful in the skin lesions and through contact with other humans continues its journey to other people.
The infection begins in upper respiratory mucosa cells, spreads to the lymph and the tonsils, and, then, is carried by T cells in the blood to the skin where it causes a widespread skin rash. The virus is, also, able to enter sensory neurons, take over transport motors and travel to the ganglion cell bodies. It then completely changes behavior and becomes dormant in the neuron for years. When it awakens, varicella, again, travels on the axon microtubule transport system to the skin to cause the painful zoster rash in the region that neurons innervate. The virus is plentiful in the skin lesions and through contact with other humans continues its journey to other people.
Structure and Lifestyle
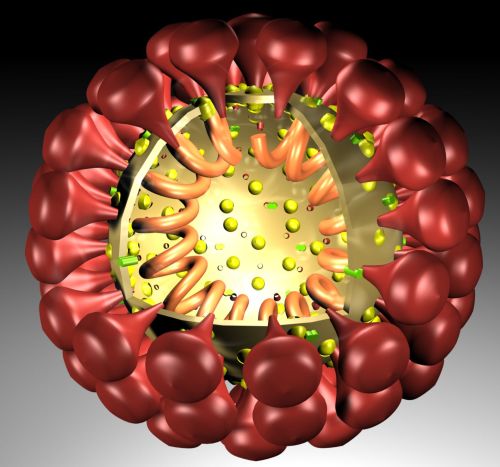
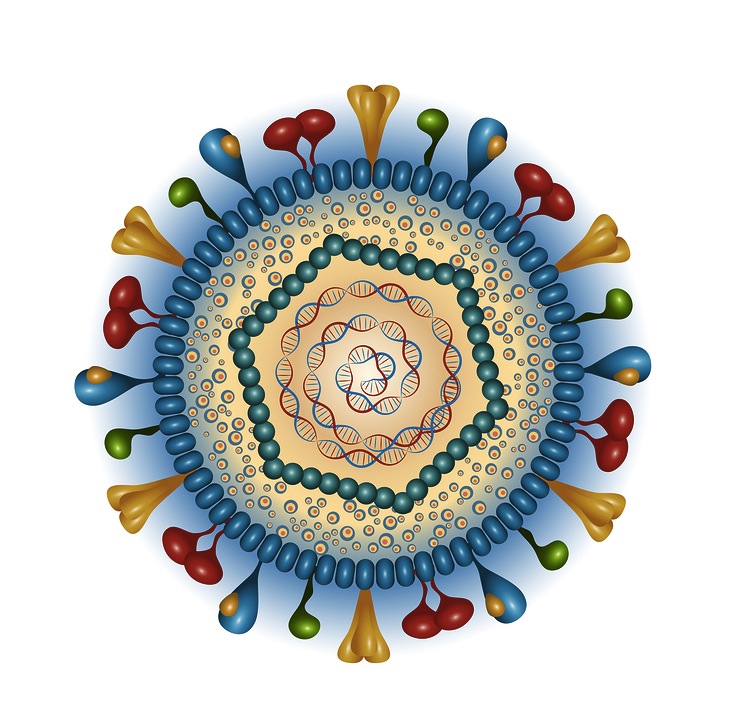 The outer layer is a lipid membrane envelope with the fusion complex that allows it to grab onto and fuse with human cell’s membranes. The next layer is the tegument with many essential proteins including the critical IE immediate early release protein. The hard capsid beneath is an icosahedron with 20 triangular faces, 30 edges and 12 vertices, which is about 100 nm in diameter.
The outer layer is a lipid membrane envelope with the fusion complex that allows it to grab onto and fuse with human cell’s membranes. The next layer is the tegument with many essential proteins including the critical IE immediate early release protein. The hard capsid beneath is an icosahedron with 20 triangular faces, 30 edges and 12 vertices, which is about 100 nm in diameter.
The viruses envelope is high in fatty molecules stolen from human cellular membranes. The virus inserts a large number of glycoproteins into the membranes—the  glycoproteins are manufactured using the host cell’s machinery.
glycoproteins are manufactured using the host cell’s machinery.
Inside the envelope there is a layer of proteins, called tegument, which the virus uses for regulation of its linear double stranded DNA. These proteins are DNA promotors. They are released when the virus enters the cell and the envelope is removed by the cell.
The first entry into the cell is not totally understood, but varicella uses five distinct receptors to fuse with the skin cell’s membrane, allowing DNA to enter the cell. This is assumed to be through fusion with the cell’s membrane or by a sac that is brought into the cell. There are many complex glycoproteins that interact with the cell surface and allow this fusion.
One major protein that is released when the coat is removed is IE62 (immediate early protein 62), which is a transcription factor to activate viral material in the nucleus. This protein knows to travel to the nucleus even before the DNA gets there. Manufacturing more virus depends upon the virus transcription factors that act on both the human DNA and the viral DNA. Several of the 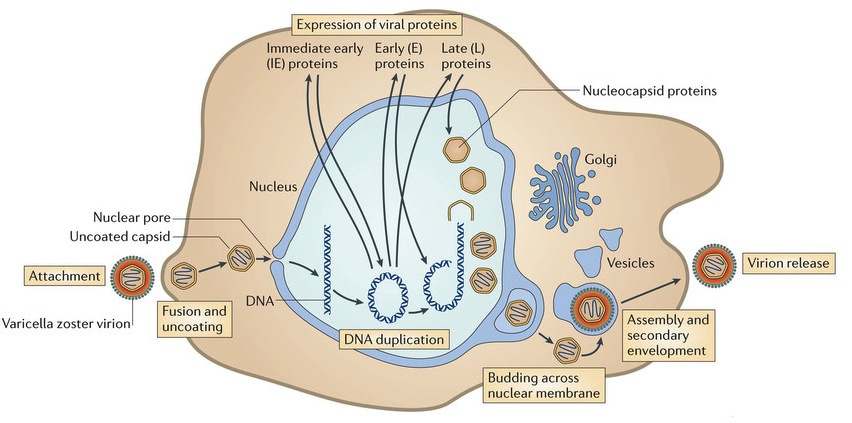 proteins serve as kinases to activate transcription factors. The IE62 molecule combines with many human molecules to form a large complex that regulates the process.
proteins serve as kinases to activate transcription factors. The IE62 molecule combines with many human molecules to form a large complex that regulates the process.
The harder protein capsid, beneath the membrane-like envelop and the tegument, docks by the nuclear pore at the cell’s membrane. The virus DNA is then injected into the nuclear pore. This capsid fuses with the nuclear membranes and awaits a new virus from the nucleus where it disengages from the nuclear membrane, covers the virus and travels to the outside cell membrane. The virus coated with the capsid goes to the cell’s Golgi, where it is dressed with its original protein layer and a membrane envelop for its journey. The entire virus is then released from the cell as if it were a sac filled with neurotransmitters.
The surface molecules are so similar to the human cell surface that in this process some human cells combine into a syncytium, which includes the material from many cells with multiple nuclei. Stimulating this syncytium allows varicella to infect more cells. This entire process takes about 9 hours.
Varicella’s Genome
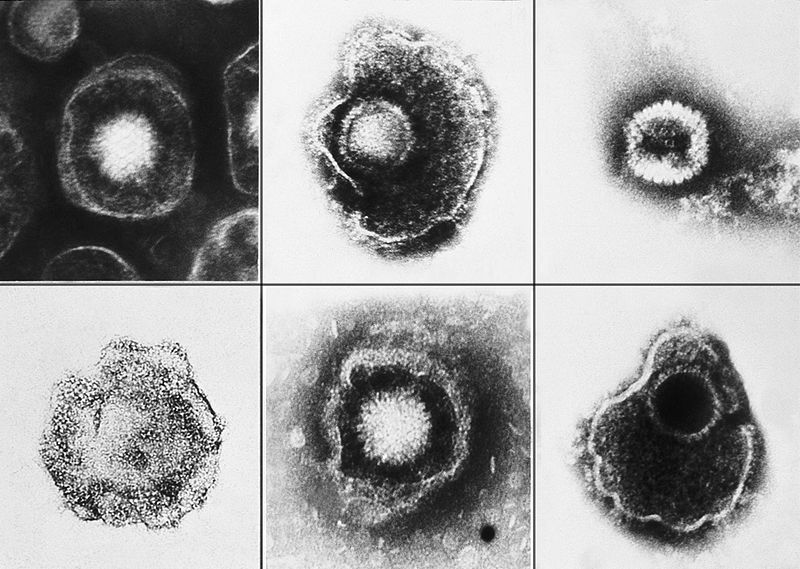 Varicella’s DNA includes at least 71 active regions, which can make proteins, with many promotor regions associated with each. The DNA is divided into one long region and one short region. The most important proteins have duplicate DNA regions. Varicella’s DNA can exist in linear or circular form. Of these 71 regions, 40 genes, including 8 glycoproteins, appear to be critical for all herpes viruses; these 40 genes are involved in DNA copying, DNA cutting and packaging, metabolism of nucleic acids, and capsid building. Proteins for replication include RNA reductase, DNA polymerase, DNA binding protein, DNA replication origin binding protein, protein kinases, as well as other DNA related proteins.
Varicella’s DNA includes at least 71 active regions, which can make proteins, with many promotor regions associated with each. The DNA is divided into one long region and one short region. The most important proteins have duplicate DNA regions. Varicella’s DNA can exist in linear or circular form. Of these 71 regions, 40 genes, including 8 glycoproteins, appear to be critical for all herpes viruses; these 40 genes are involved in DNA copying, DNA cutting and packaging, metabolism of nucleic acids, and capsid building. Proteins for replication include RNA reductase, DNA polymerase, DNA binding protein, DNA replication origin binding protein, protein kinases, as well as other DNA related proteins.
In T cells
 Specific T cells are essential to clear a primary infection and avoid reactivation from latency.
Specific T cells are essential to clear a primary infection and avoid reactivation from latency.
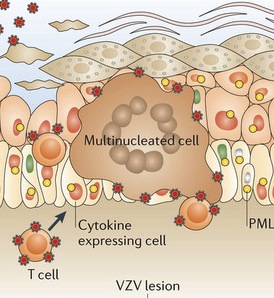
However, varicella is able to fool the very intelligent T cells and multiply inside of them. The T cell populations CD3, CD4 and CD8 and the dual CD4CD8 allow varicella to travel and multiply inside. These T cells pick up the virus in lymph nodes near the respiratory mucosa. Varicella, also, can infect dendritic cells. The T cells with varicella are attracted specifically to skin cells because of the signaling proteins and receptors on the T cells. Varicella spreads near hair follicles because of the many small blood vessels around the follicles. The T cells, traveling in the blood, can transmit across these small blood cell walls.
Remarkably, the virus induces specific signals and factors that keep the T cells alive and gives them time to transport the virus. Because of these varicella stimulated factors, the T cells do not fuse together, which means that each T cell is infected individually. The virus controls the T cell fusion in order to maintain them as transport in and out of the tissues. The ability to infect so many T cells allows the widespread skin lesions. In the skin because of varicella stimulation, cells do fuse together to form a syncytium with a large amount of the virus present—this allows increased spread of the virus throughout the skin region.
A glycoprotein in varicella’s short DNA section is essential for T cell infection. These factors stop the T cell from killing itself (since it normally would if infected) and counteract other cytokines like IFN, which might stop the infection. A critical component of the virus action on T cells decreases the expression of MHC1, which is the primary way that other cells would know the T cell was infected.
In Skin Cells
 The skin lesions are formed by innate immune responses from the cell. The lesions take two to three weeks to form. They appear near hair follicles where skin cells have fused together into a syncytium with many nuclei and many viruses. Complex immune factors and cytokines are involved in the reaction. The T cell reaction only occurs later after the skin lesions have formed. The major reaction is innate and not adaptive, which allows for an infection that is not catastrophic and allows the long-term existence of the virus in the tissues. The vaccine eliminates most of this reaction.
The skin lesions are formed by innate immune responses from the cell. The lesions take two to three weeks to form. They appear near hair follicles where skin cells have fused together into a syncytium with many nuclei and many viruses. Complex immune factors and cytokines are involved in the reaction. The T cell reaction only occurs later after the skin lesions have formed. The major reaction is innate and not adaptive, which allows for an infection that is not catastrophic and allows the long-term existence of the virus in the tissues. The vaccine eliminates most of this reaction.
Varicella is able to suppress the innate response to allow skin lesions with many live viruses for transmission to other humans. The skin cells release the newly minted 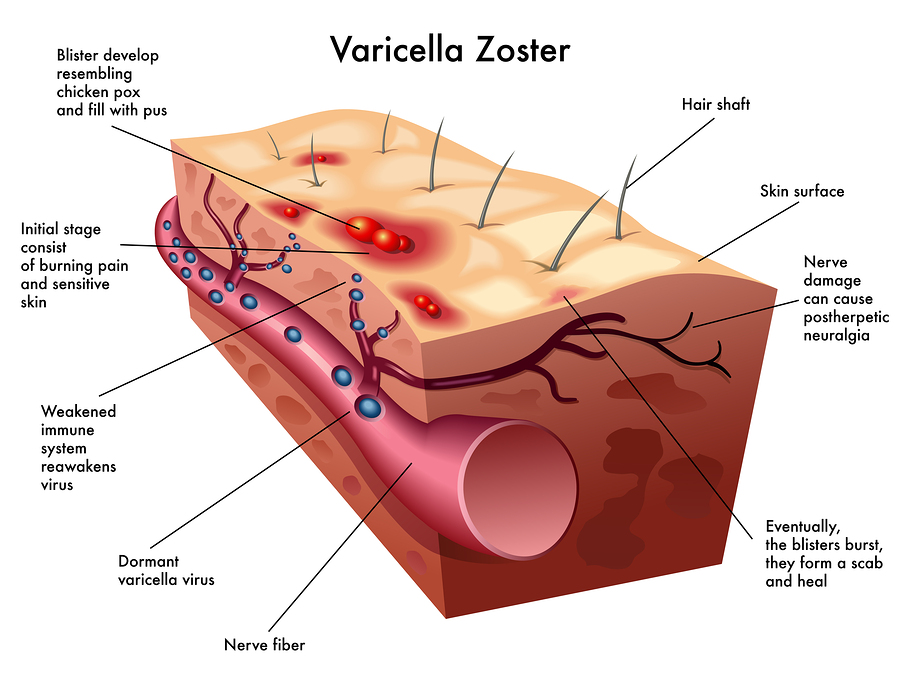 varicella. Varicella uses specific proteins that interfere with the immune factor IFN. It, also, alters cell signaling to stop cell death pathways to allow continued infection. Some virus further manipulates this pathway to produce cancers. Varicella uses another tricky technique to keep the cell alive longer by stimulating autophagosomes that compete with cell death pathways. By competing with the cell death pathways it maintains the longevity of the cell and, therefore, the longevity of varicella in the cell.
varicella. Varicella uses specific proteins that interfere with the immune factor IFN. It, also, alters cell signaling to stop cell death pathways to allow continued infection. Some virus further manipulates this pathway to produce cancers. Varicella uses another tricky technique to keep the cell alive longer by stimulating autophagosomes that compete with cell death pathways. By competing with the cell death pathways it maintains the longevity of the cell and, therefore, the longevity of varicella in the cell.
 A very significant anti nuclear viral protein PML is usually stimulated by the immune factor IFN. Normally, PML forms cages to trap the virus in the cells nucleus by grabbing its capsid. PML usually forms an important structure in the nucleus called the PML nuclear body for this purpose. One of the virus proteins, ORF61, binds to the PML with SUMO and stops the PML function (see post on SUMO – SUMO is a series of molecules that tag other molecules and alter signaling pathways – the previous post described the war between virus and the host cell using ubiquitin and SUMO.)
A very significant anti nuclear viral protein PML is usually stimulated by the immune factor IFN. Normally, PML forms cages to trap the virus in the cells nucleus by grabbing its capsid. PML usually forms an important structure in the nucleus called the PML nuclear body for this purpose. One of the virus proteins, ORF61, binds to the PML with SUMO and stops the PML function (see post on SUMO – SUMO is a series of molecules that tag other molecules and alter signaling pathways – the previous post described the war between virus and the host cell using ubiquitin and SUMO.)
The virus can spread without fusion of cells, but in the skin it uses this fusion to spread. The virus is able to alter important signaling pathways that preserve the cell’s membrane. It makes it easier to not have to deal with many barriers. The virus only allows the fusion to occur in the outermost layer of the skin. A special section of the virus genome appears to affect both T cells and skin cells at the same time in these very different complex ways.
In Neurons
 When first infected, neurons have detectable virus for 4 to 8 weeks before latency. During latency up to 10 varicella are present in 4% of the neurons in the dorsal route ganglion, but protein factors are not produced during this residence. When the virus is activated, 25% of the neurons are involved with the production of many measurable protein factors.
When first infected, neurons have detectable virus for 4 to 8 weeks before latency. During latency up to 10 varicella are present in 4% of the neurons in the dorsal route ganglion, but protein factors are not produced during this residence. When the virus is activated, 25% of the neurons are involved with the production of many measurable protein factors.
In T cells and skin cells there are adaptive immune responses, but there aren’t any in neuron infection. This occurs because varicella specifically silences genes in the neurons with interfering RNAi. Critical to this silencing of neurons is that varicella’s capsid can be collected and sequestered by PML bodies in the neuron and satellite cells’ nucleus. Cages of the PML fibers contain virus and newly forming virions in the neurons that are infected.
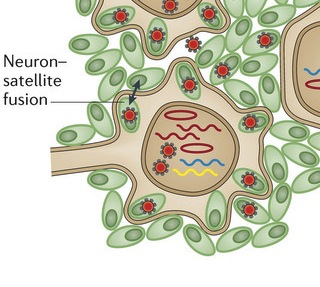 A very unusual action of the virus is to stimulate neurons and satellite cells (the cells that surround peripheral neurons) to fuse in the same way as skin cells. These neuron-satellite cells become a syncytium with multiple nuclei and many viruses. This fusion spreads the virus further in the ganglion, which, also, increases the area of skin that is involved in the painful lesions. These changes do not reverse easily, which is a source of ongoing pain after zoster infection.
A very unusual action of the virus is to stimulate neurons and satellite cells (the cells that surround peripheral neurons) to fuse in the same way as skin cells. These neuron-satellite cells become a syncytium with multiple nuclei and many viruses. This fusion spreads the virus further in the ganglion, which, also, increases the area of skin that is involved in the painful lesions. These changes do not reverse easily, which is a source of ongoing pain after zoster infection.
Varicella Doesn’t Just Ride on Motor But Commandeers It
 When the herpes virus rides on the dynein motor travelling on the microtubule transportation system in the neuron’s axon, it doesn’t passively take the ride. In keeping with its general intelligence, it commandeers the dynein motor and alters the ATP energy mechanism. In this process it, actually, speeds up the motor transport to arrive much more quickly at the nucleus in one trip and then later the second trip back to the skin. In fact, herpes can travel in pieces or as a whole virus in vesicles. The return trip uses a second mechanism.
When the herpes virus rides on the dynein motor travelling on the microtubule transportation system in the neuron’s axon, it doesn’t passively take the ride. In keeping with its general intelligence, it commandeers the dynein motor and alters the ATP energy mechanism. In this process it, actually, speeds up the motor transport to arrive much more quickly at the nucleus in one trip and then later the second trip back to the skin. In fact, herpes can travel in pieces or as a whole virus in vesicles. The return trip uses a second mechanism.
The Remarkable Intelligent Varicella Virus
 Varicella exhibits an enormous amount of intelligent actions, including many very complex signals to induce fusion of skin cells, neurons and satellite cells, and to avoid fusion of T cells. These actions are all aimed to maintain the cells needed for varicella’s lifestyle. These signals specifically avoid programmed cell death to maintain the cells as varicella vehicles.
Varicella exhibits an enormous amount of intelligent actions, including many very complex signals to induce fusion of skin cells, neurons and satellite cells, and to avoid fusion of T cells. These actions are all aimed to maintain the cells needed for varicella’s lifestyle. These signals specifically avoid programmed cell death to maintain the cells as varicella vehicles.
Varicella uses five distinct receptors to fuse with the skin cell’s membrane when entering the cell. It fools the very intelligent T cell and travels inside of it. It stimulates specific signals that maintain the T cell and stop the cell’s possible fusion in order for varicella to continue to travel. It foils the complex nuclear pore mechanisms while entering the nucleus of each type of cell. Using very complex machinery it forms circular DNA and uses the cell’s genetic process to reproduce. In the skin cell it uses complex signaling to cause a skin cell syncytium and to stop programmed cell death. The new virus leaves the skin cell and enters a sensory neuron. It hijacks the complex dynein transport motor, taking it over, accelerating it and directing it the long way up the axon to the neuron’s cell body and nucleus. With different tricks it fools the neuron’s nuclear pore complex. Once in the neuron’s nucleus it alters its own behavior and becomes quiescent. It silences neuronal genes using RNAi. It manufactures specific molecules that stop further activity in the neuron’s nucleus so as not to kill the neuron. It can remain there for many years without interfering with function. At some time it awakens and leaves the nucleus, again travelling back on the microtubule machinery along the long axon. It, then, leaves the neuron, enters skin cells where it reproduces and uses this vantage point to jump to another person.
These sequential maneuvers are each very complex. How does this small virus with DNA for 70 proteins know how to orchestrate this complex lifestyle? How can we not be impressed with the intelligence of the remarkable varicella virus?