 Previous posts have described intelligent behavior in cells, organelles, and microbes. Viruses and jumping genes, which are virtually just a strand of DNA or RNA, also show complex behavior. Since DNA and RNA are involved in the process of reproduction and creation of new molecules, there is an intuitive understanding how viruses and jumping genes might engage in intelligent behavior. But, can a protein alone engage in intelligent behavior? Is a prion an intelligent protein?
Previous posts have described intelligent behavior in cells, organelles, and microbes. Viruses and jumping genes, which are virtually just a strand of DNA or RNA, also show complex behavior. Since DNA and RNA are involved in the process of reproduction and creation of new molecules, there is an intuitive understanding how viruses and jumping genes might engage in intelligent behavior. But, can a protein alone engage in intelligent behavior? Is a prion an intelligent protein?
A prion is a protein that can take different shapes and in one particular structure recruits others to change into this form as well. These proteins then clump together into a large mass that disrupts and kills neurons. It is a 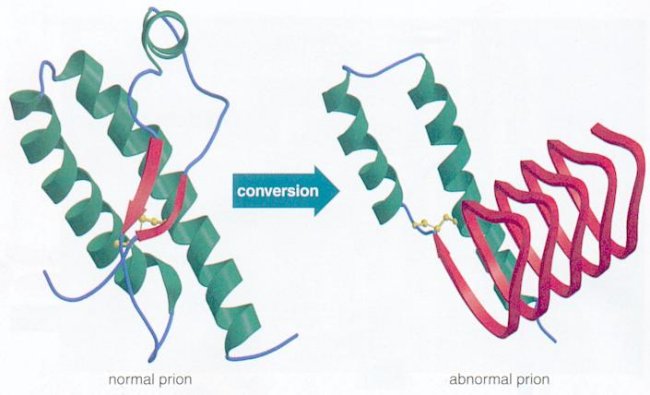 version of a protein that can perpetuate growth like a crystal. When the protein is in the brain and sits in a neuronal membrane, a signal is created that can kill cells. It also kills cells by enlarging masses of protein clumps.
version of a protein that can perpetuate growth like a crystal. When the protein is in the brain and sits in a neuronal membrane, a signal is created that can kill cells. It also kills cells by enlarging masses of protein clumps.
The prion protein, alone, becomes a pathogen that creates infections. Although it is not absolutely certain that the protein has no help from a virus, current evidence appears to show that it acts alone without any genetic material. The disease in humans is called Creutzfeldt-Jakob disease. There are many different animal versions, as well. Some, like Mad-Cow disease comes from eating infected meat.
There is now increasing evidence that the clumping of proteins with prion  like behavior is a more general phenomenon and important proteins that are involved in many of the major brain diseases have prion like behavior—Alzheimer’s, Parkinson’s, Huntington’s, and ALS.
like behavior is a more general phenomenon and important proteins that are involved in many of the major brain diseases have prion like behavior—Alzheimer’s, Parkinson’s, Huntington’s, and ALS.
Five of these critical proteins have both normal and abnormal effects and several appear to work together in disease processes. Also, research demonstrates that neurons secrete prion like proteins in vesicles (like neurotransmitters) and that other neurons pick up these vesicles. Very recently, it was observed that prion like proteins, such as amyloid beta, attach and clump on neuronal receptors consisting of prions and material from the extra cellular matrix (see Extra Cellular Matrix post.) It was, also, shown that prion like proteins break open vesicles inside neurons, escape, and attack mitochondria.
The basic protein that becomes a prion has multiple important normal roles in cells, particularly in immune cells. Also, the immune system is now known to be critical to its spread. A simple protein that has only “self” antigens falls beneath the cracks of the immune surveillance system. In fact, it can create prion crystals in the immune system. Like a virus, it hijacks components of the immune system and rides along with immune cells into the central nervous system.
Normal Function of the Prion Protein
 The prion protein is known as PrP before it becomes the pathogen PrPsc. PrP has many different normal important physiological roles. PrP is involved in maintaining myelin formation, development of T cells and is critical in stem cells that make blood cells and M cells of the intestine immune centers. PrP is involved in virus reproduction and modulation of inflammation in the CNS.
The prion protein is known as PrP before it becomes the pathogen PrPsc. PrP has many different normal important physiological roles. PrP is involved in maintaining myelin formation, development of T cells and is critical in stem cells that make blood cells and M cells of the intestine immune centers. PrP is involved in virus reproduction and modulation of inflammation in the CNS.
PrP can modulate the immune response to its own advantage. Prp can inhibit the function of macrophages, which engulf or phagocytize other debris.
When the PrP becomes a prion its normal function stops. It is possible that it is this loss of normal function that causes some of the damage in prion diseases, not just the accumulation of the clumped proteins.
Change of the Protein to a Prion
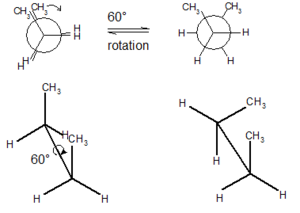 Prion proteins can exist in multiple structures called conformers. These different shapes are created by sigma bonds in their chemical structures that allow the rotation of parts of the protein.
Prion proteins can exist in multiple structures called conformers. These different shapes are created by sigma bonds in their chemical structures that allow the rotation of parts of the protein.
The normal protein that becomes a prion has either alpha helixes or disordered structures (see post on Protein Folding). This shape is found in most cells and does not cause any damage. The prion version of the same protein has beta sheets, a completely different structure. The new prion protein forms a clump and becomes a base, which draws in other protein building a crystal mass. It is not yet clear exactly how this transformation occurs, but most cellular folding (see post on folding) is managed by complex large proteins called chaperones. It appears that lipids act as the chaperones for prions.
 Protein folding is based upon the amino acid sequence. Because of the different three-dimensional shapes of each amino acid, the folding of a protein with 400 amino acids is extremely complex. In a previous post it was noted that current science is unable to calculate the folding of even an average sized protein using all of the world’s supercomputers—it would take a billion years. Yet, when cells, including microbes (and even viruses), edit their own genes and alternatively splice messenger RNA to make specialized new proteins, they appear to know the shape it will take when it folds. This folding needs the chaperone molecules and the folding takes place in a millisecond.
Protein folding is based upon the amino acid sequence. Because of the different three-dimensional shapes of each amino acid, the folding of a protein with 400 amino acids is extremely complex. In a previous post it was noted that current science is unable to calculate the folding of even an average sized protein using all of the world’s supercomputers—it would take a billion years. Yet, when cells, including microbes (and even viruses), edit their own genes and alternatively splice messenger RNA to make specialized new proteins, they appear to know the shape it will take when it folds. This folding needs the chaperone molecules and the folding takes place in a millisecond.
Prions Enter
Prions can enter the body through food and injection. Injection of prions has occurred during corneal transplants. It was originally discovered in Kuru, a prion disease that was self injected by people using sticks for crushing animal brains to eat. They sharpened their sticks by stabbing themselves injecting the prion into their blood. Prions can, also, enter through blood transfusions, which demonstrates that blood allows prions entry into the brain. If prions are on the skin or in aerosols, they also end up in lymph tissue.
 Prions can enter orally and cross the wall of the GI tract. They survive gastric acid and intestinal enzymes. The prion seems to enter through specialized immune tissue in the intestine called the Peyer’s patch. In experiments with animals, inflammation of the bowel increases the number of prions entering. M immune cells in the Peyer’s patch take samples of the material floating by for immune surveillance. M cells pick up prions from and bring them into the body. There are other possible mechanisms where intestinal cells actively transport the proteins into the cell.
Prions can enter orally and cross the wall of the GI tract. They survive gastric acid and intestinal enzymes. The prion seems to enter through specialized immune tissue in the intestine called the Peyer’s patch. In experiments with animals, inflammation of the bowel increases the number of prions entering. M immune cells in the Peyer’s patch take samples of the material floating by for immune surveillance. M cells pick up prions from and bring them into the body. There are other possible mechanisms where intestinal cells actively transport the proteins into the cell.
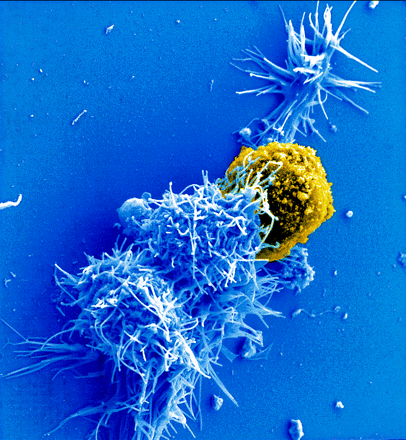 Dentritic cells are critical immune cells that pick up pieces of material throughout body including in the blood stream and lymphatic vessels. The material is then presented by dendritic cells to a mature T cell in the lymph nodes. Dendritic cells can, also, pick up prions and transfer them into the lymph system to the lymph nodes.
Dentritic cells are critical immune cells that pick up pieces of material throughout body including in the blood stream and lymphatic vessels. The material is then presented by dendritic cells to a mature T cell in the lymph nodes. Dendritic cells can, also, pick up prions and transfer them into the lymph system to the lymph nodes.
Immune System Harbors Prions
In naturally occurring diseases of many wild animals, prions multiply in the lymph tissues before entering the brain. Tonsil biopsies can pick up prion disease. The spleen is a major place for prions, especially the stromal region. The spleen appears to be critical for early multiplication. It appears that T cells are not responsible for prion multiplication. B cells 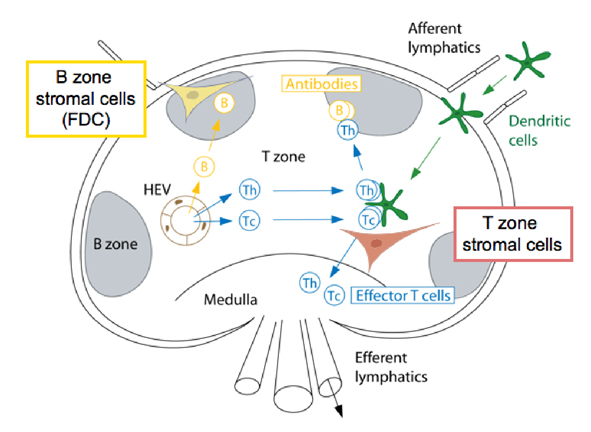 are necessary for prions to enter the brain, but once they are in the brain they are not dependent on B cells to spread.
are necessary for prions to enter the brain, but once they are in the brain they are not dependent on B cells to spread.
While B cells make considerable amount of prion precursors, they are not essential for prion multiplication. The follicular dendritic cells do appear to be critical by trapping debris, including prions, and allowing them to multiply. Because prions start as natural proteins they do not appear to trigger the ordinary innate immune response that responds to unusual shapes including cancer cells. They also don’t trigger the adaptive immunity of T cells and antibodies.
Inflammation and Prions
Prions reproduce and grow in the lymph tissues including tonsils, spleen, Peyer’s patches in the intestine and lymph nodes. In lymph tissue, prions are associated with follicular dendritic dells. Follicular dendritic cells capture  pieces of pathogens marked for destruction by complement. It appears that some of the complement molecules are necessary for prion development.
pieces of pathogens marked for destruction by complement. It appears that some of the complement molecules are necessary for prion development.
Dendritic cells can increase the number of prions even in non-lymph tissue when there is chronic inflammation with lymphocytes present. One example is kidney infection, where prions are found in the urine before any symptoms are noted. In animals, prions occur in mastitis, mammary glands, and in colostrum and milk of animals. Prions in milk might be a prominent route in sheep. Chronic inflammation can modify prion disease by enlarging the areas that they are found.
In the Brain
 Prions accumulate in peripheral lymph tissues, which are innervated by sympathetic nerves. Most of the time prions spread into the brain through the autonomic nervous system. In experiments where sympathetic nerves were cut this stopped the spread.
Prions accumulate in peripheral lymph tissues, which are innervated by sympathetic nerves. Most of the time prions spread into the brain through the autonomic nervous system. In experiments where sympathetic nerves were cut this stopped the spread.
Prions enter nerves that are closest to the lymph tissues and spread from there. But, it is not clear how. A variety of possible routes include direct cell-to-cell contact, transmission between cells in vesicles like neurotransmitters, tunneling nanotubes and free floating protein particles. Prions could  be transported along microtubules.
be transported along microtubules.
It is now known that neurons expel and pick up vesicles with prion like proteins. A receptor has been found related to tau and amyloid beta.
Once in brain, prions spread along neuronal circuits. The clump becomes a template for conformation change of the protein and this alteration is communicated to the next protein, then to the next neuron.
Do Immune Cells Stop Prions?
They might slightly, but, not enough. Previous posts have discussed how special pattern recognition receptors, known as toll-like receptors (TLR), trigger immune responses in the innate immune response. TLR do seem to protect somewhat against prions, but they don’t trigger the ordinary innate response or the adaptive response from T cells. There are no PrP antibodies noted in infected people or animals. The lymph tissue was abnormal but those with and without critical cytokines, such as IL-6, had the same rate of growth of the prion.
As the prion accumulates in the brain it causes wide areas of damage called spongiform encephalopathy, which includes neuro-inflammation with many more activated astrocytes and microglia.
Microglia and Prions
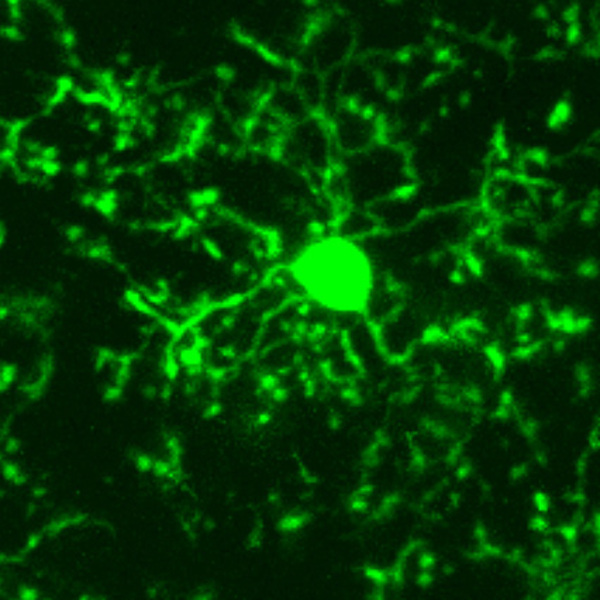 There is evidence that microglia can slow the accumulation of prions, but they can also do the opposite. It appears that there must be communication between the microglia and the astrocytes for the protection to be effective. But, despite a clear response from microglia, prion disease destroys brains. In fact, during the battle with prions the microglia alter their cell type from M1, which fights inflammation, to the pro inflammatory M2 version of the microglia. M2 actually spread the prion further.
There is evidence that microglia can slow the accumulation of prions, but they can also do the opposite. It appears that there must be communication between the microglia and the astrocytes for the protection to be effective. But, despite a clear response from microglia, prion disease destroys brains. In fact, during the battle with prions the microglia alter their cell type from M1, which fights inflammation, to the pro inflammatory M2 version of the microglia. M2 actually spread the prion further.
As prion accumulates in the brain many cytokines are stimulated, which increase inflammation—IL-1, TNF and IL-6. But, at the same times anti-inflammatory cytokines are also increased—IL-4 and IL-10 in CSF. Anti nuclear factor, NF-kappa-beta is stimulated, which causes neuronal apoptosis (cell death).
Other Possible Prions
 Several other protein molecules are now suspected to be prions. These are critical molecules in the major degenerative diseases of the nervous system—Alzheimer’s, Parkinson’s, ALS, and Huntington’s. In fact, some of these appear to work together.
Several other protein molecules are now suspected to be prions. These are critical molecules in the major degenerative diseases of the nervous system—Alzheimer’s, Parkinson’s, ALS, and Huntington’s. In fact, some of these appear to work together.
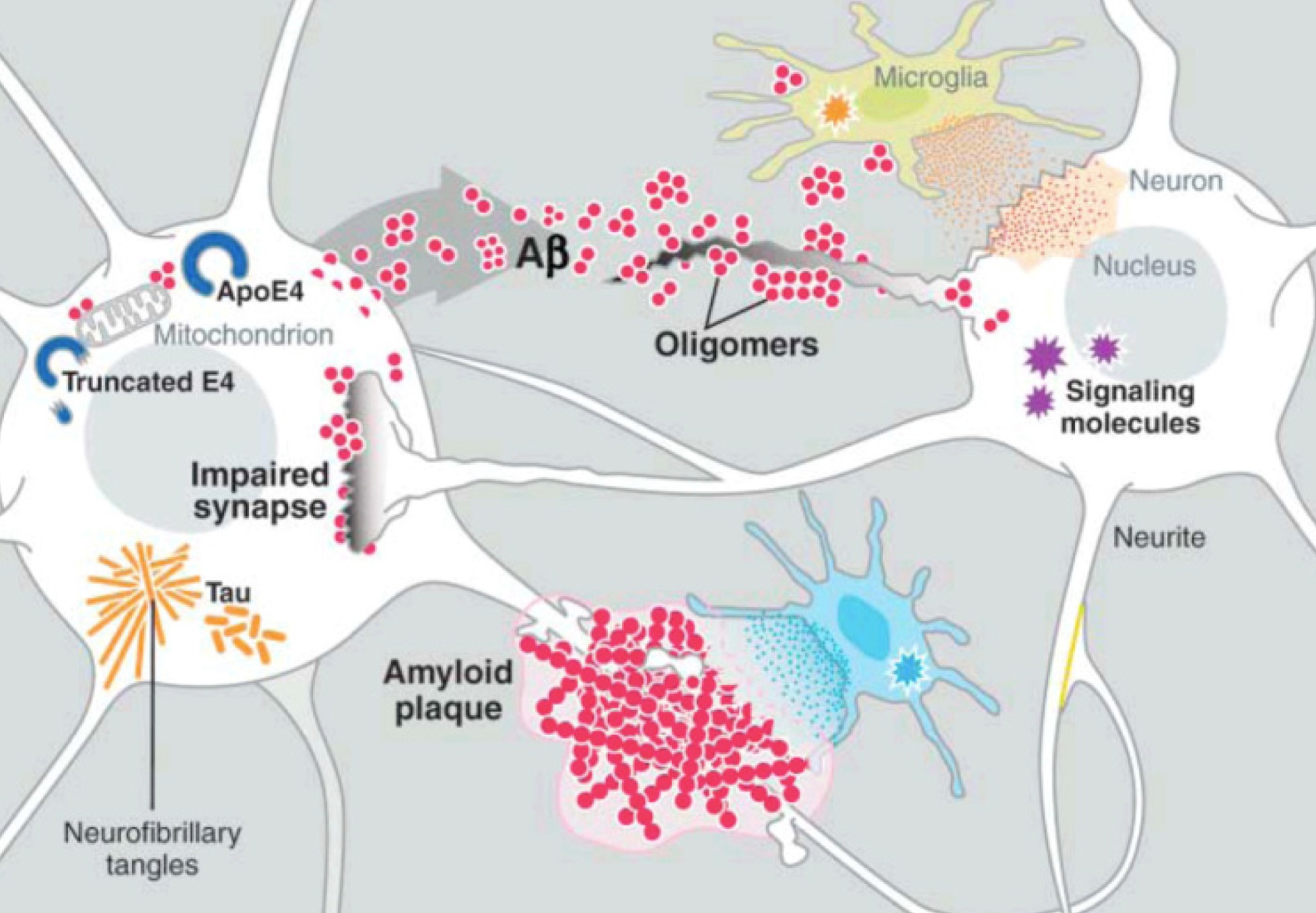
Five critical molecules have been studied in this regard—amyloid and tau in Alzheimer’s, alpha-synuclein in Parkinson’s, SOD1 in ALS, and polyglutamine in Huntington’s. Just like PrP each have normal functions in the brain and, then, when misfolded, become prions. It appears that amyloid beta, tau and alph-synuclein influence each other in Alzheimer’s and Parkinson’s.
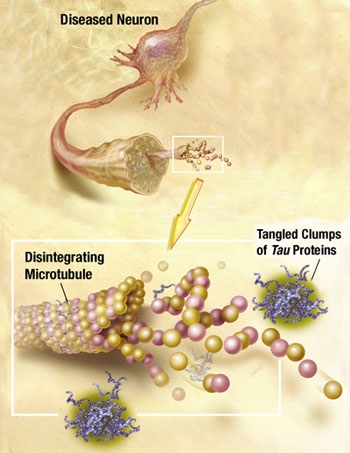 Recently, it has been discovered that tau can be released from neurons and taken up by the next neuron. Alpha synuclein can also be transmitted out of a neuron in vesicles, enter another cell, and then when inside the cell break out of the vesicle disrupting the mitochondria. Very recently a receptor has been found that allows the early clumping of amyloid beta in Alzheimer’s. While the details remain to be discovered, all of the prion functions are already in place—clumping, which is transmitted out of one cell and brought into another to form more clumps.
Recently, it has been discovered that tau can be released from neurons and taken up by the next neuron. Alpha synuclein can also be transmitted out of a neuron in vesicles, enter another cell, and then when inside the cell break out of the vesicle disrupting the mitochondria. Very recently a receptor has been found that allows the early clumping of amyloid beta in Alzheimer’s. While the details remain to be discovered, all of the prion functions are already in place—clumping, which is transmitted out of one cell and brought into another to form more clumps.
This activity is reminiscent of virus activity. But, a virus is made of DNA or RNA. These prions are just a protein molecule.
 It is also interesting that all of these diseases are mostly sporadic (80%) meaning they start without a particular cause. A smaller number are either through exposure or genetic mutations. All of these proteins are important parts of multiple different varied diseases. This occurs because there are multiple different ways the clumps can form. For example, amyloid is involved in Alzheimer’s, myositis, and angiopathy. Abnormal tau and alpha-synuclein both cause multiple different diseases. Also, in all of the diseases, symptoms appear after many years.
It is also interesting that all of these diseases are mostly sporadic (80%) meaning they start without a particular cause. A smaller number are either through exposure or genetic mutations. All of these proteins are important parts of multiple different varied diseases. This occurs because there are multiple different ways the clumps can form. For example, amyloid is involved in Alzheimer’s, myositis, and angiopathy. Abnormal tau and alpha-synuclein both cause multiple different diseases. Also, in all of the diseases, symptoms appear after many years.
Evidence of Spreading
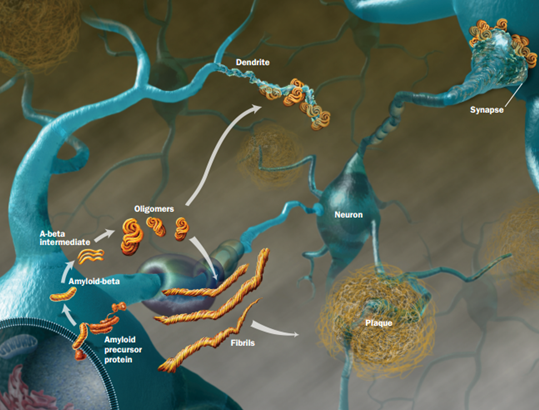 All of these prion like proteins appear to spread along brain circuits. Tau is known to start in the entorhinal region near the nose spreading to entorhinal cortex along glutamate neurons, then to hippocampus, amygdala and neocortex.
All of these prion like proteins appear to spread along brain circuits. Tau is known to start in the entorhinal region near the nose spreading to entorhinal cortex along glutamate neurons, then to hippocampus, amygdala and neocortex.
Prions introduced through surgery can spread from the eye along visual pathways. Spread can occur along the vagus nerve to medulla, pons, midbrain. In transplants for treatment of Parksinson’s, the new neurons develop the same disease from the local cells. Injection of abnormal amyloid beta, tau and alpha synuclein in animal experiments shows spread into the new brain.
Alzheimer’s Prions
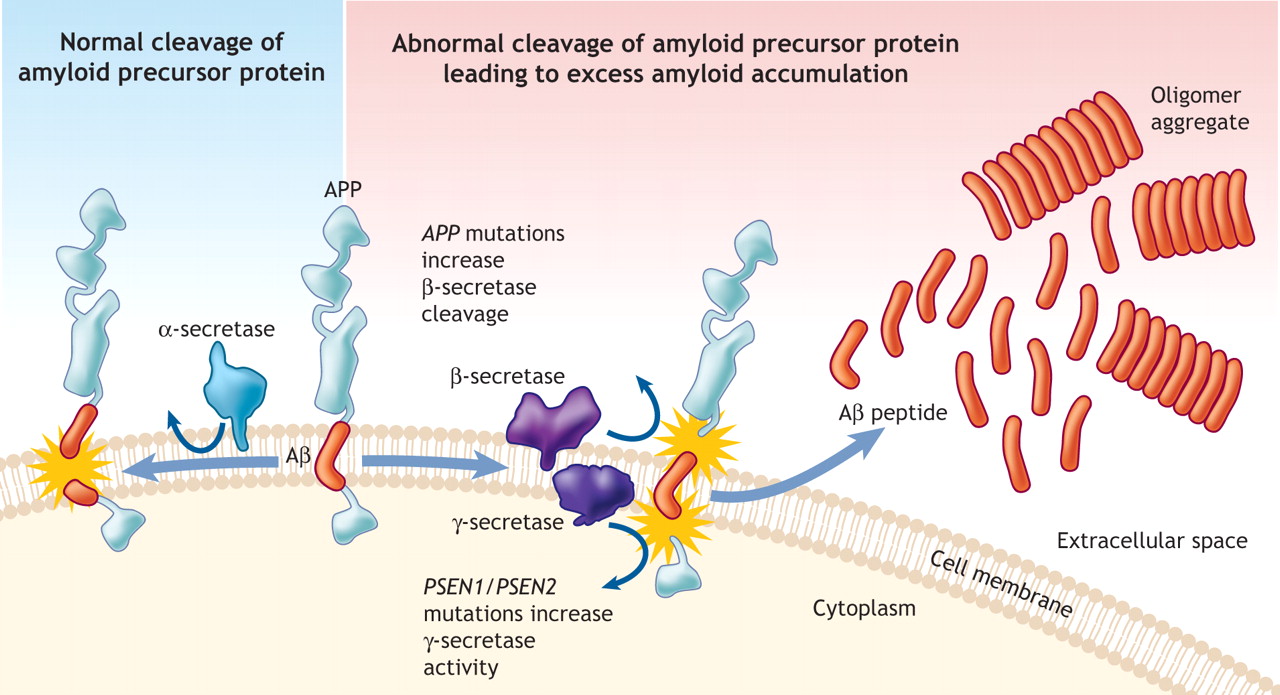
There are many connections between PrP, and the critical proteins amyloid beta, tau, and alpha-synuclein. Prions appear to serve as a receptor for amyloid beta to start plaques.
 Amyloid beta is a peptide (small protein) of approximately 40 amino acids. Normally, amyloid is not toxic and is involved in protection against oxidation, regulation of cholesterol transport, and serves as a transcription factor. It is cleared by the glymaphatic system (see ECM post).
Amyloid beta is a peptide (small protein) of approximately 40 amino acids. Normally, amyloid is not toxic and is involved in protection against oxidation, regulation of cholesterol transport, and serves as a transcription factor. It is cleared by the glymaphatic system (see ECM post).
Abnormally, it folds and forms clumps, fibers and plaques. Like the prion protein it can take different forms in clusters. One important type is an amyloid beta oligomer. When small molecules combine into a long chain it is called a polymer, when it is a small chain is an oligomer.
Amyloid beta oligomers might be the toxic agents. But, oligomers appear to need tau for this process. Tau normally ties microtubules together in the neuron. Recently, it was shown that amyloid beta oligomers interact with the PcP binding to it.
A previous post, Extra Cellular Matrix, described a receptor in the extra cellular matrix made from one of the critical extra cellular proteoglycans. This receptor is called HSPG, or heparin sulfate proteoglycan. HSPG appears to be part of the Alzheimer mechanism.
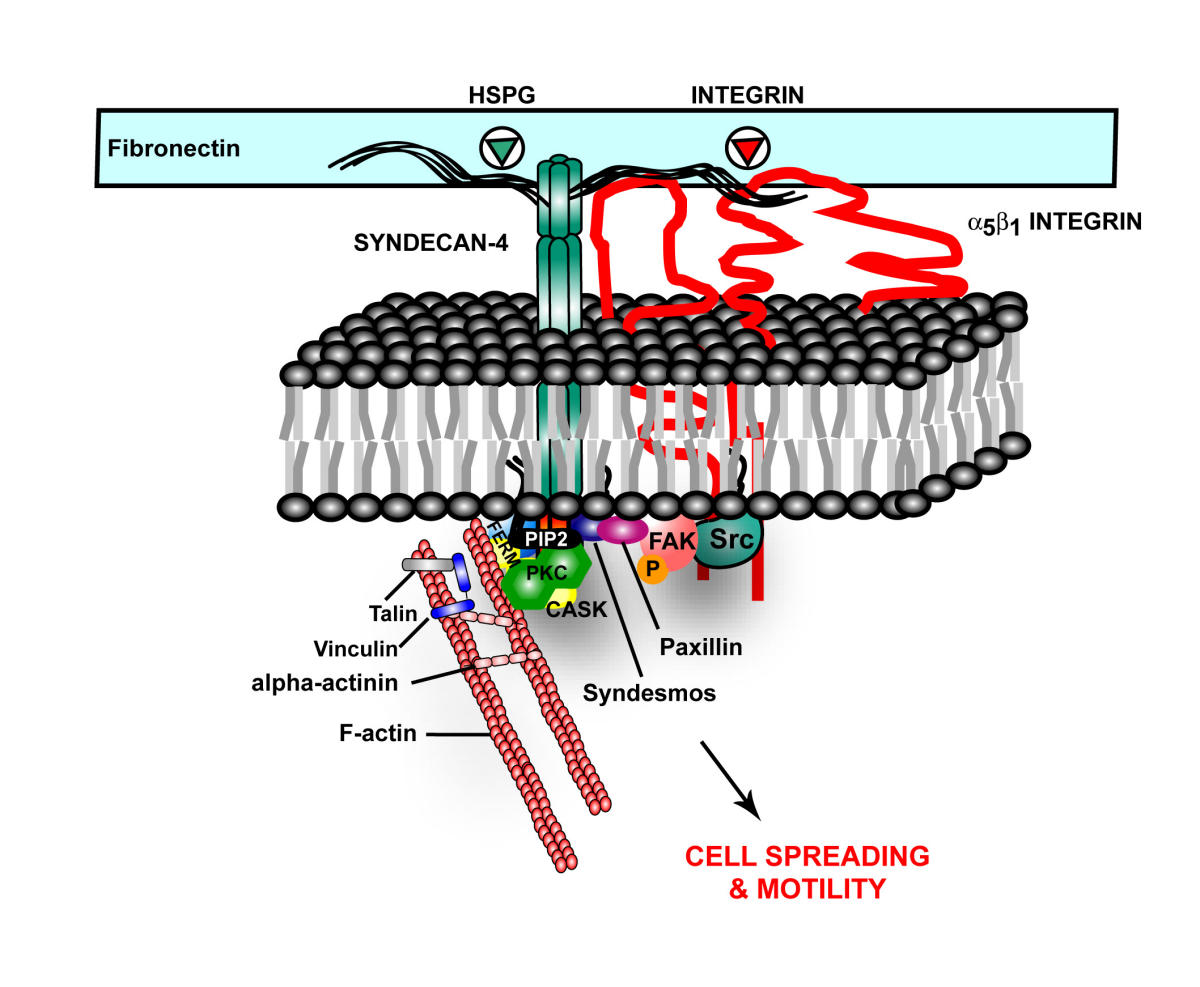 In Alzheimer’s, tau misfolds and sticks into clumps. HSPG binds the tau, bring it into the cell. It also appears that HSPGs are essential for cell to cell spread of alpha-synuclein.
In Alzheimer’s, tau misfolds and sticks into clumps. HSPG binds the tau, bring it into the cell. It also appears that HSPGs are essential for cell to cell spread of alpha-synuclein.
Also, HSPG, is part of amyloid plaques and neurofibrillary tangles. HSPGs appear to increase the formation of amyloid beta by stopping the enzymatic cleavage of the amyloid peptide and also interacting with apolipoprotein E (APOE), which is known as a major risk factor for Alzheimer’s. APOE helps transport lipid in the brain and helps with repair of neurons. The lipoprotein molecules include lipids that are used for the maintenance of neurons. When APOE4 cannot bind to HSPG then the lipoprotein function is impaired.
Is A Prion An Intelligent Protein
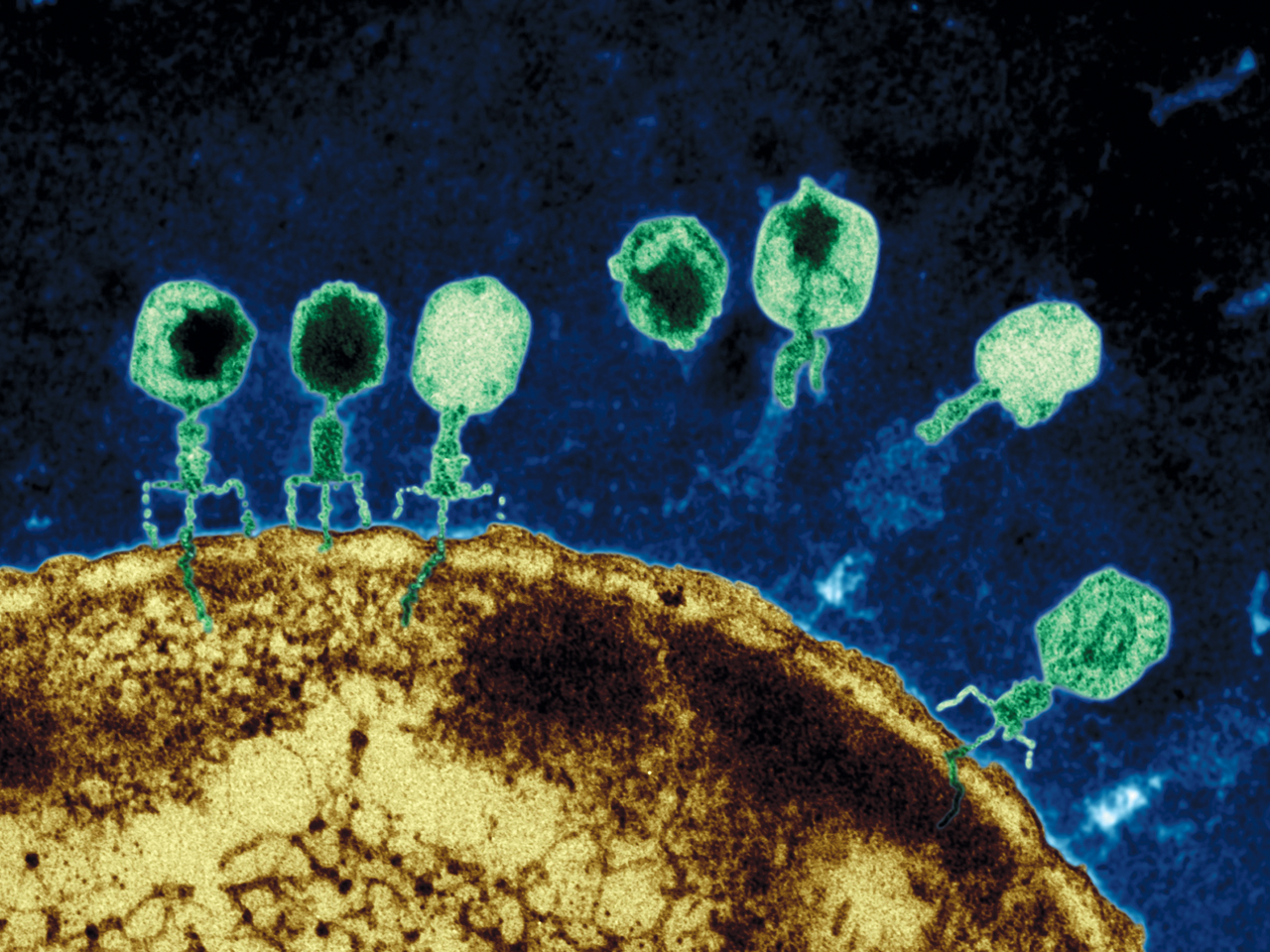 Viruses, and virus like particles such as jumping genes, demonstrate complex behavior. A virus will perform a sequence of a dozen coordinated complex actions, each involving seeming intelligence. An example is the herpes virus—enters skin cell, fools nuclear pore, creates DNA machinery to reproduce, leaves cell to enter neuron, hijacks complex transport system, fools new nuclear pore, changes behavior to be quienscent, re activates, and again hijacks the transport system.
Viruses, and virus like particles such as jumping genes, demonstrate complex behavior. A virus will perform a sequence of a dozen coordinated complex actions, each involving seeming intelligence. An example is the herpes virus—enters skin cell, fools nuclear pore, creates DNA machinery to reproduce, leaves cell to enter neuron, hijacks complex transport system, fools new nuclear pore, changes behavior to be quienscent, re activates, and again hijacks the transport system. 
Prions are an even simpler infectious agent. While there is much less known about them than viruses, they also engage in complex behavior—exiting and entering of cells, foiling the immune system, multiplying and transporting themselves with immune cells or along neuron circuits and recruiting other proteins to form large structures.
It is only in the past several years that the full extent of virus behavior has been observed. Prions are still hard to study and more will be known about them in the next several years. Is it possible that we will add proteins to the list of living entities with intelligence and complex behavior?