 With vast brain complexity, the cause of Alzheimer’s is not clear and there are no current treatments that alter the disease process. Most Alzheimer’s researchers assume the cause is abnormal clumps of amyloid-β that kill neurons. Some define early pre clinical Alzheimer’s as those with increased amyloid-β and are testing medications in those with this increase without Alzheimer’s.
With vast brain complexity, the cause of Alzheimer’s is not clear and there are no current treatments that alter the disease process. Most Alzheimer’s researchers assume the cause is abnormal clumps of amyloid-β that kill neurons. Some define early pre clinical Alzheimer’s as those with increased amyloid-β and are testing medications in those with this increase without Alzheimer’s.
But, in fact, this theory may not be correct. There are cases with a large amount of amyloid and no Alzheimer’s and the opposite—people with Alzheimer’s and few amyloid clumps. The research is very complex and it is possible that amyloid clumps are an effect and not a cause.
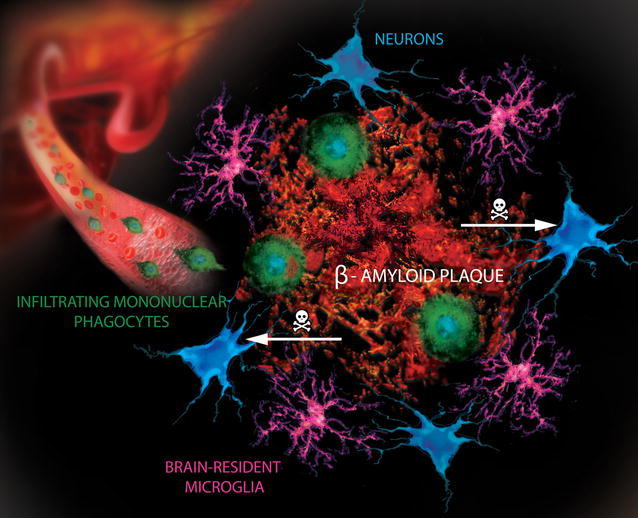 The intense focus on only one theory has decreased funding for other theories, which are part of a very complex story with many different types of findings at the same time. Recently, the question of another mis folded protein tau has been raised as a possible cause. Abnormal tau is connected with amyloid in animal models. Other important theories include inflammation in the brain with abnormal microglia. Another is the unusual prion like properties of all of the major misfolded proteins—amyloid-β, tau, α-synuclein, SOD1, TDP43 and polyglutamines—which are part of Alzheimer’s, Parkinson’s, ALS and Huntington’s. These mis folded proteins are actually transmitted by neurons in circuits mostly at synapses and with at times with exosomes.
The intense focus on only one theory has decreased funding for other theories, which are part of a very complex story with many different types of findings at the same time. Recently, the question of another mis folded protein tau has been raised as a possible cause. Abnormal tau is connected with amyloid in animal models. Other important theories include inflammation in the brain with abnormal microglia. Another is the unusual prion like properties of all of the major misfolded proteins—amyloid-β, tau, α-synuclein, SOD1, TDP43 and polyglutamines—which are part of Alzheimer’s, Parkinson’s, ALS and Huntington’s. These mis folded proteins are actually transmitted by neurons in circuits mostly at synapses and with at times with exosomes.
There are many other possible causes of Alzheimer’s including oxidative, DNA or mitochondrial damage; failure of autophagy and lysosomes; loss of calcium homeostasis; excess excitement from glutamate; failure of the neuron cell cycle; fungal disease; and inflammation caused by neurons (see post on neuro inflammation).
Previous posts about Alzheimer’s have highlighted the great complexity of the possible mechanisms involved in the disease, including its relation to inflammation and the inverse relationship with cancer through the p53 system.
This post will discuss the current controversy about whether amyloid-β is a cause or an effect of Alzheimer’s.
Alzheimer’s Subtypes

There are definitely different types of Alzheimer’s. But, it is not clear how many. Two have been described—early familial and late sporadic—but, the common late variety might have multiple subtypes. Recent research shows that the fibers of amyloid take many different shapes at the molecular level, called polymorphisms. The complex process of protein folding into sheets and then larger structures based on the specific small fiber structure determines these different types of clumps. Different types of amyloid fibers form enlarging clumps of the same type and are self-propagating. These different fiber subtypes may produce different clinical pictures and produce Alzheimer subtypes.
One researcher recently described what he thinks are three types: inflammatory with increased c-reactive protein and albumin; non inflammatory with other metabolic changes; and cortical in younger people with loss of language not memory first.
A known autosomal dominant gene causes early onset familial type and the much more frequent late onset type does not have a clear cause. Amyloid-β peptide—known as Aβ—is known to be the cause of the early onset dominant type. The assumption for the later onset type is amyloid triggering a cascade of reactions that causes the clumping of abnormal amyloid.
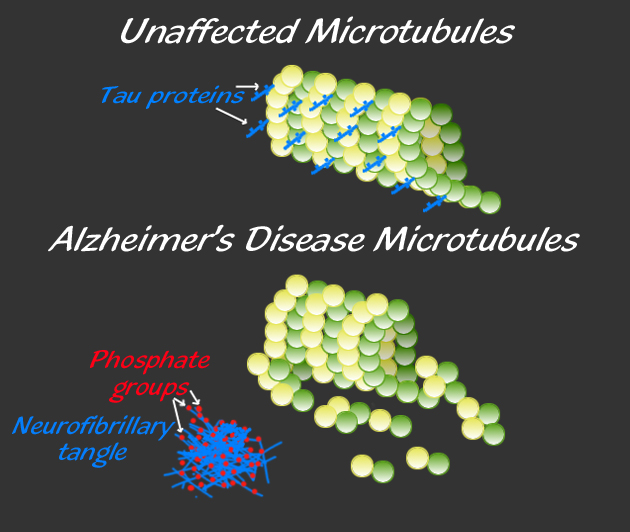
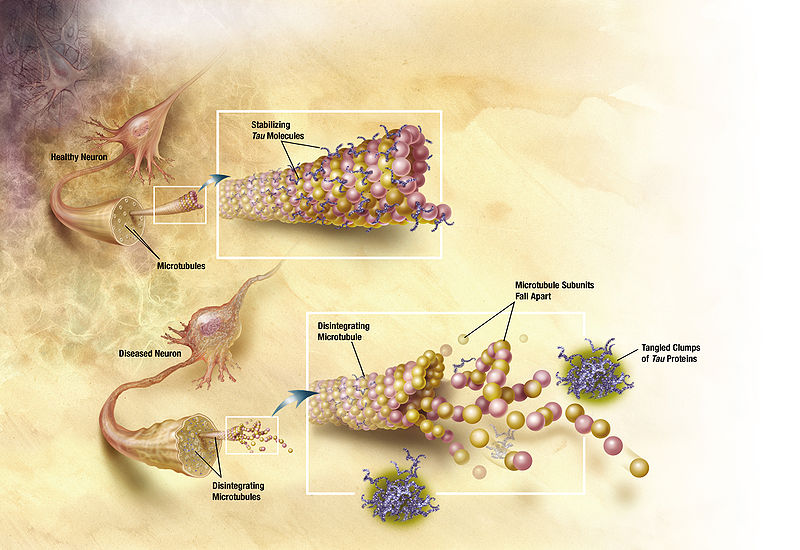 The late onset type includes clumps of tau, which also damages neurons. Tau is a protein, which holds together the structure of microtubules, and is altered by the addition of a phosphate. When altered, tau forms clumps and causes “neurofibrillary tangles,” one of the two microscopic hallmarks of the disease (the other being amyloid plaques). Some evidence supports the fact that Aβ specifically causes the Alzheimer brain damage in this late onset type. But, even the data in favor of Aβ as causative conclude that more than just Aβ is necessary to produce the symptoms, such as tau. Conversely, another brain disease—cerebral amyloid angiopathy—only needs Aβ.
The late onset type includes clumps of tau, which also damages neurons. Tau is a protein, which holds together the structure of microtubules, and is altered by the addition of a phosphate. When altered, tau forms clumps and causes “neurofibrillary tangles,” one of the two microscopic hallmarks of the disease (the other being amyloid plaques). Some evidence supports the fact that Aβ specifically causes the Alzheimer brain damage in this late onset type. But, even the data in favor of Aβ as causative conclude that more than just Aβ is necessary to produce the symptoms, such as tau. Conversely, another brain disease—cerebral amyloid angiopathy—only needs Aβ.
Genetics of Alzheimer’s
 The familial type of Alzheimer’s is caused by a mutation in three genes that are parts of the amyloid Aβ manufacturing process. APP is the gene that makes the amyloid precursor protein, which is cut to become Aβ. Genes PSENI1 and PSENI2 make part of the enzyme that cuts the amyloid precursor protein producing Aβ. These genes produce the two subunits of the γ-secretase enzyme—presenilin 1 and 2. When all three mutations occur, there is a greater production of the Aβ plaques. When there are two APP genes on chromosome 21 there is, also, greater manufacture of the Aβ plaques. Because Downs’ syndrome people have an extra chromosome 21 (trisomy) there is more plaque made as well.
The familial type of Alzheimer’s is caused by a mutation in three genes that are parts of the amyloid Aβ manufacturing process. APP is the gene that makes the amyloid precursor protein, which is cut to become Aβ. Genes PSENI1 and PSENI2 make part of the enzyme that cuts the amyloid precursor protein producing Aβ. These genes produce the two subunits of the γ-secretase enzyme—presenilin 1 and 2. When all three mutations occur, there is a greater production of the Aβ plaques. When there are two APP genes on chromosome 21 there is, also, greater manufacture of the Aβ plaques. Because Downs’ syndrome people have an extra chromosome 21 (trisomy) there is more plaque made as well.
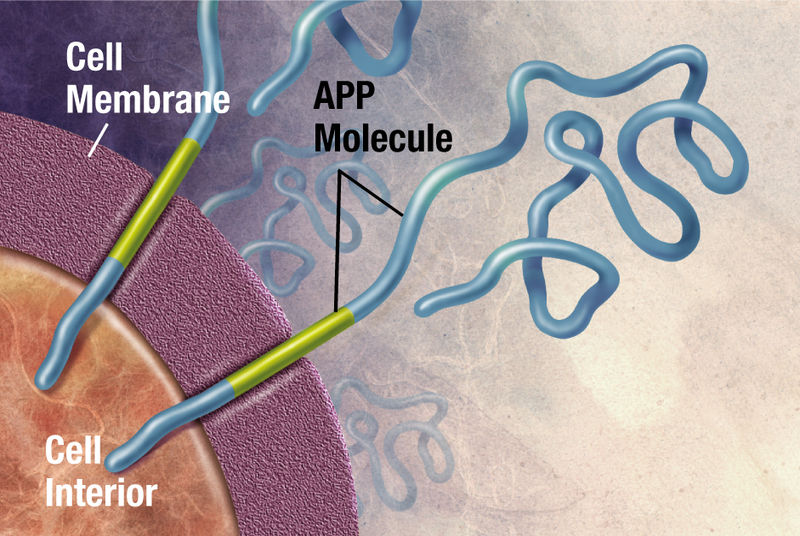 There are, in fact, a variety of different mutations with slightly different effects, but all are related to making more Aβ. The mechanisms can involve β-secretase enzyme as well, and, also, can affect the ability of the cell to clean up Aβ. These varied mechanisms demonstrate that it is the increased clumping ability and decreased clearance ability that are most important for more plaques.
There are, in fact, a variety of different mutations with slightly different effects, but all are related to making more Aβ. The mechanisms can involve β-secretase enzyme as well, and, also, can affect the ability of the cell to clean up Aβ. These varied mechanisms demonstrate that it is the increased clumping ability and decreased clearance ability that are most important for more plaques.
In all of these mechanisms, tau abnormalities appear later. In fact, a new Alzheimer model in a dish showed that tau neurofibrillary tangles would inevitably come from Aβ plaques.
Sporadic Late Onset Alzheimer’s is the Common Form
 The cases that are not clearly inherited are called sporadic. There are no easy genetic answers for this type. APOE is the most important risk factor, but is a highly complex gene.
The cases that are not clearly inherited are called sporadic. There are no easy genetic answers for this type. APOE is the most important risk factor, but is a highly complex gene.
APOE, or apolipoprotein E, is a class of apolipoproteins (see post) that is an essential part of triglyceride metabolism. It is produced by the liver and by macrophage white blood cells. It is critical for the metabolism of cholesterol. APOE is produced by astrocytes in the brain and transports cholesterol to neurons. This transport triggers APOE receptors.
 There are four types of APOE and the ε4 type increases the chance of getting Alzheimer’s. The ε2 has the least chance. When people have one ε4, the risk is 4x and if both chromosomes have it then the risk is 12x.
There are four types of APOE and the ε4 type increases the chance of getting Alzheimer’s. The ε2 has the least chance. When people have one ε4, the risk is 4x and if both chromosomes have it then the risk is 12x.
In fact, APOE has many different effects in the brain. It affects amyloid clumping in the CSF, especially the variety of Aβ42, which the most toxic type of Aβ (more than Aβ40 the less toxic type). A new very rare mutation has recently been identified of APP that lowers Aβ and decreases Alzheimer’s.
Tau in Alzheimer’s
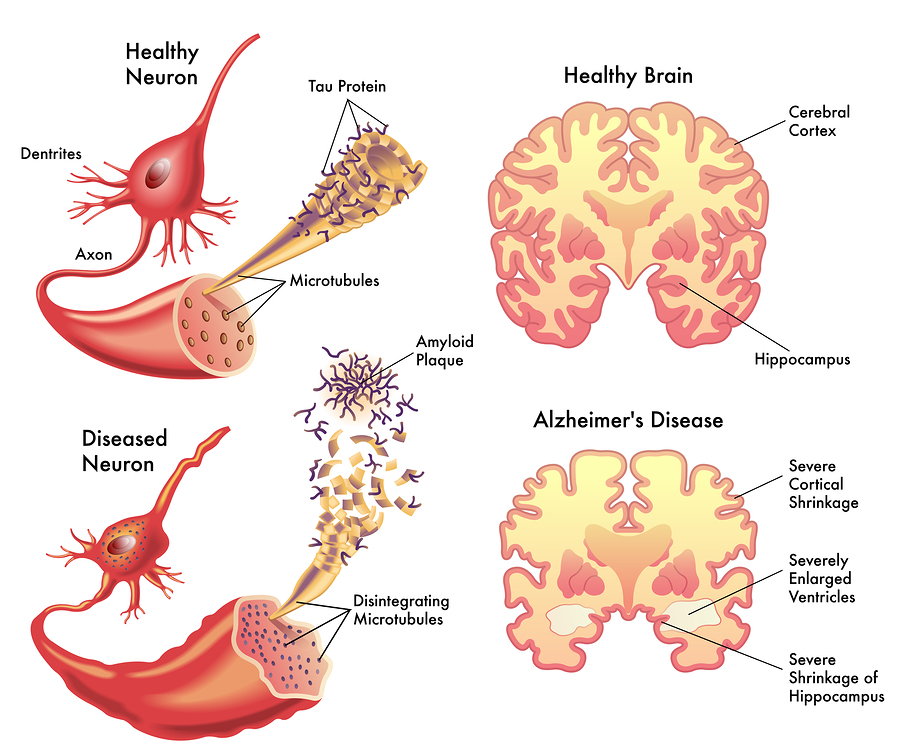 Tau proteins normally stabilize microtubules. See posts on the critical importance of microtubules for brain function. Tau exists mostly in neurons, but is sometimes seen in other types of cells. It exists in small amounts in astrocytes and oligodendrocytes. Many different tau proteins are produced by alternative splicing from one gene (MAPT-microtubule-associated protein tau) on chromosome 17. Abnormal tau occurs in both Alzheimer’s and in Parkinson’s disease.
Tau proteins normally stabilize microtubules. See posts on the critical importance of microtubules for brain function. Tau exists mostly in neurons, but is sometimes seen in other types of cells. It exists in small amounts in astrocytes and oligodendrocytes. Many different tau proteins are produced by alternative splicing from one gene (MAPT-microtubule-associated protein tau) on chromosome 17. Abnormal tau occurs in both Alzheimer’s and in Parkinson’s disease.
One of the major arguments against the amyloid hypothesis is that the symptoms do not correlate with increased plaque and happen many years later. A previous post documented that abnormal proteins of the type in Alzheimer’s and Parkinson and ALS are transmitted from neuron to neuron along circuits. This transmission is along the neuronal circuits and the transmission takes place at the synapse. The transmission from neuron to neuron defines the types of regions that will have the clumps. In Alzheimer’s disease, plaque starts in the frontal and pre cuneus brain regions. But, neuronal damage occurs in different regions—the entorhinal cortex and hippocampus, memory centers.
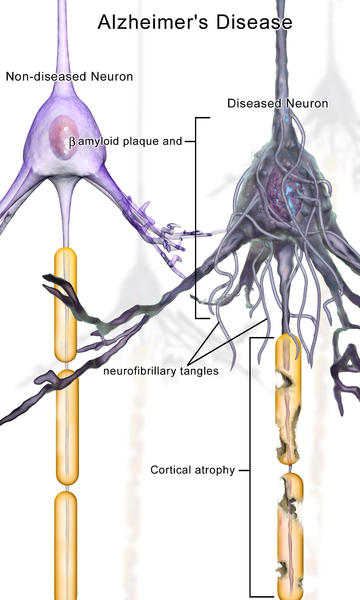
It is abnormal tau that is much more correlated with the disease symptoms and the specific regions that are damaged. Abnormal tau is transported in neuronal circuits in the opposite direction as the Aβ plaques. The regions that receive the tau clumps correlates more with the damage that is seen the disease. The Aβ plaques are not near the prominent damage. One hypothesis is that amyloid drives tau damage. The same regions and progression of damage occur in the familial and the sporadic and in both there is a difference between amyloid regions and the damaged regions.
Tau is phorphorylated to become abnormal and exists before amyloid in the brainstem and entorhinal cortex. In all normal elderly people, tau fibrillary tangles exists in limbic regions without any cognitive symptoms. When there are Aβ problems, then tau spreads into the cortex causing symptoms. The tau pathology seems to only occur when there is Aβ plaque pathology at the same time but in different regions.
Normal elderly have tau phosphorylation in the hippocampus with no neuronal damage. When Aβ exists in this region there are dementia symptoms. Tau mutations, also, do not cause Aβ. This data is consistent with Aβ triggering tau pathology and Alzheimer’s symptoms. In research where Aβ is added to a culture it eventually causes tau abnormalities and neuron damage. Another study found that altering the β and γ-secretase enzyme blocked the tau pathology, showing a connection.
Another finding is that Aβ can stimulate other abnormal proteins such as α-synuyclein, the protein that when abnormal causes Parkinson’s. α-synuclein appears, also, in Alzheimer’s.
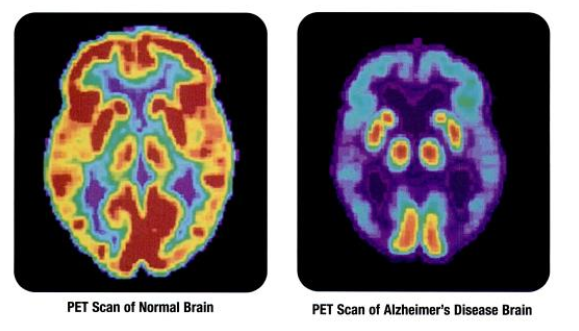 Many elderly have Aβ plaques and no Alzheimer’s. But, those with Aβ plaques and no tau yet have a higher probability of getting the disease in the next 2 to 7 years. The follow up studies have yet to be done to be accurate in these predictions. If there is evidence of brain injury, then the risk is higher. (The theory behind this effect of brain injury is the transmission of abnormal proteins from brain injury regions to other regions). Some studies show that markers for Aβ and then markers for tau pathology both exist years before cognitive changes occur.
Many elderly have Aβ plaques and no Alzheimer’s. But, those with Aβ plaques and no tau yet have a higher probability of getting the disease in the next 2 to 7 years. The follow up studies have yet to be done to be accurate in these predictions. If there is evidence of brain injury, then the risk is higher. (The theory behind this effect of brain injury is the transmission of abnormal proteins from brain injury regions to other regions). Some studies show that markers for Aβ and then markers for tau pathology both exist years before cognitive changes occur.
Another Speculation
Another finding is that other forms of amyloid oligomers (small pieces) that are not Aβ cause other types of problems than plaques. There is some suggestion that these oligomers are more related to eventual Alzheimer’s than the type that forms plaque, possibly because they influence tau. They do appear to cause synaptic damage. But, this is mainly speculation and there are many problems with this explanation of the relation of Aβ and tau pathology, not the least is that the exact type is not known.
Another possibility is that the prion like behavior of the Aβ, tau and α-synuclein are part of it. Another possibility is that Aβ affects some global function of the brain that affects tau, but this is not known, either. Inflammation is another possibility (see post).
Another recent finding is related to a different protein fragment of the amyloid precursor protein, APP. This is the amyoid precursor protein intracellular domain, or AICD. This fragment is produced along with Aβ. AICD appears to, also damage synapse communication in Alzheimer’s.
Many Different Abnormalities Exist at the Same Time
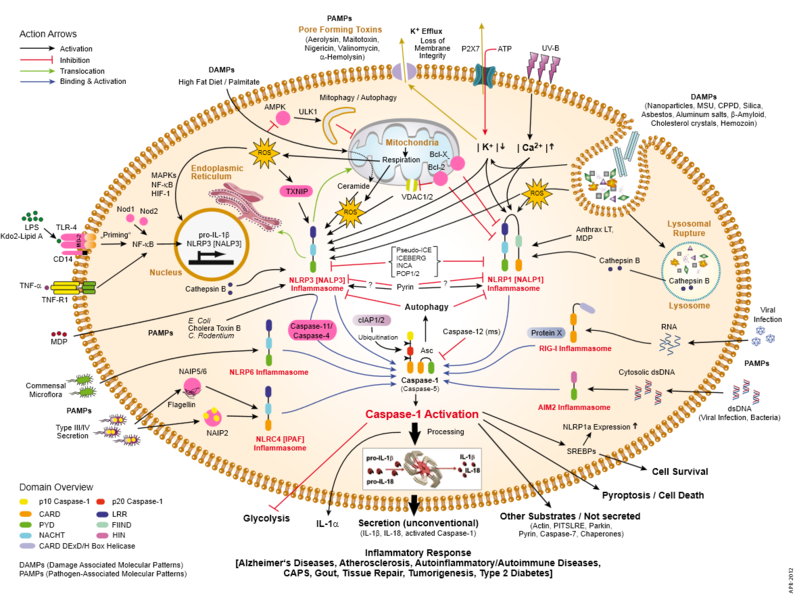
One problem with the Aβ hypothesis is that inflammation, oxidative stress, mitochondrial abnormalities and abnormal autophagy all occur in the Alzheimer’s patient. Also, vascular disease accelerates Alzheimer’s pathology as does brain injury. Recently, fungus was found in Alzheimer’s brain regions. These factors have, also, been shown to affect APP and Aβ metabolism. These factors are, also, present in many different other brain diseases. Smoking has widespread affects as well, including stimulating vascular dementia. Specific mutations affect microglia (see post)
There are, also, other fragments of APP that appear to have damaging effects like AICD. APP has many other independent brain affects that could, also, be relevant.
Another problem is that there is no clear explanation of what starts the Aβ accumulation. It could be from defective brain cleaning. There are two basic ways the brain cleans out mis folded proteins in the brain. Microglia normally clean debris from the region of the brain that each patrols.
 The second way is the newly discovered glymphatic system (glia-lymphatic.) This system of cleaning occurs by flow of fluid in the extracellular space from the region of arteries to veins and then ends up in the lymphatic system. It is remarkable that during sleep, neurons shrink by 50% so that the cleaning can be much more efficient. Sleep disruption is now shown to affect the glymphatic system that cleans abnormal protein debris in the brain. Also, sleep disruption is correlated with Alzheimer’s. (See the post Five Secrets of Brain Health).
The second way is the newly discovered glymphatic system (glia-lymphatic.) This system of cleaning occurs by flow of fluid in the extracellular space from the region of arteries to veins and then ends up in the lymphatic system. It is remarkable that during sleep, neurons shrink by 50% so that the cleaning can be much more efficient. Sleep disruption is now shown to affect the glymphatic system that cleans abnormal protein debris in the brain. Also, sleep disruption is correlated with Alzheimer’s. (See the post Five Secrets of Brain Health).
There is other evidence in favor of the amyloid hypothesis. Aβ is regulated through life and then it is disturbed including sleep and synaptic activity. Stress, also, increases Aβ. Vascular and other diseases can disrupt it. First there is Aβ then it triggers tau spreading to cortex. Then α-synuclein and inflammation set in.
Summary of Factors AGAINST Amyloid Hypothesis
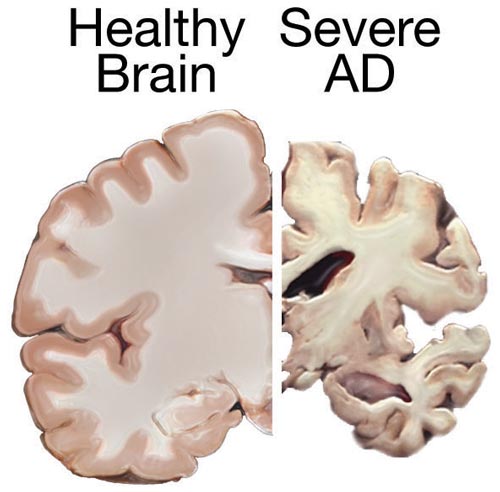 Most money and time has gone into the amyloid hypothesis, possibly at the detriment of other theories that might be equally important. There are many reasons not to believe in a simple linear affect of amyloid and Alzheimer’s.
Most money and time has gone into the amyloid hypothesis, possibly at the detriment of other theories that might be equally important. There are many reasons not to believe in a simple linear affect of amyloid and Alzheimer’s.
The Alzheimer’s brain is shrunken in the entire neo cortex and some subcortical regions and there is enlargement of the ventricles. Most of the basal nucleus (center of acetylcholine) the dorsal raphe (serotonin) and the locus ceoruleus (norepinephrine) are lost. Dopamine regions of the substantia nigra are not lost. Because of tau, it is possible it begins in the brain stem. There is, also, widespread destruction of synapses.
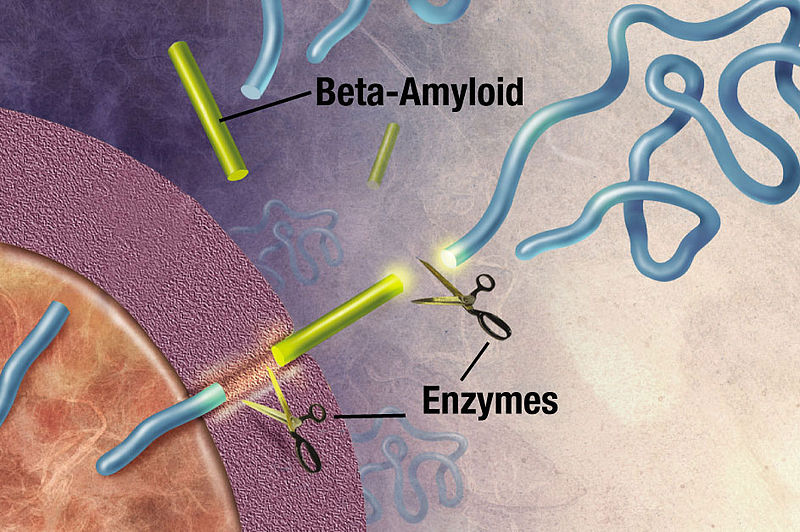
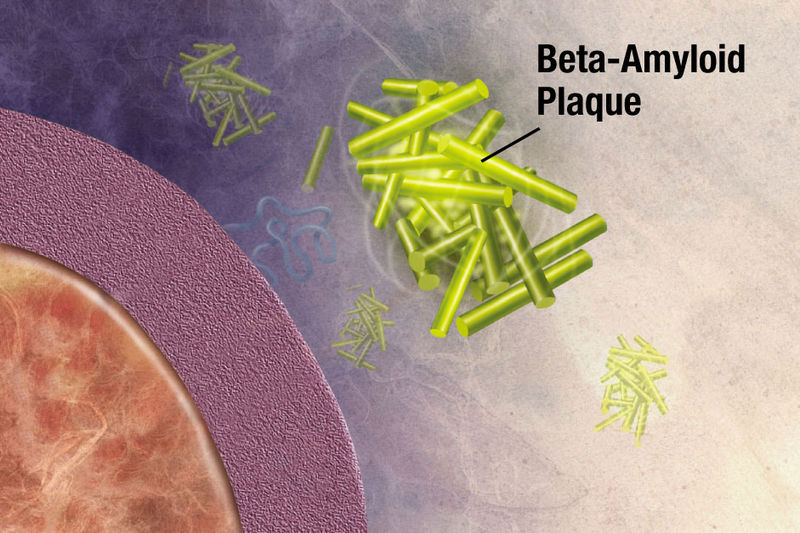
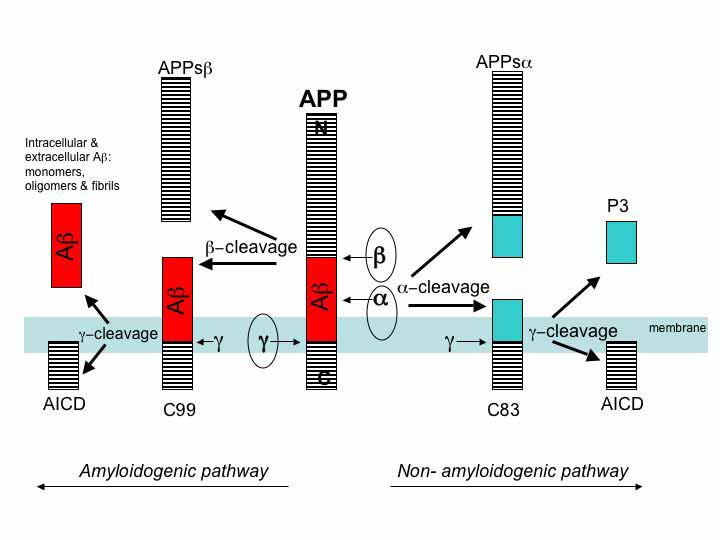
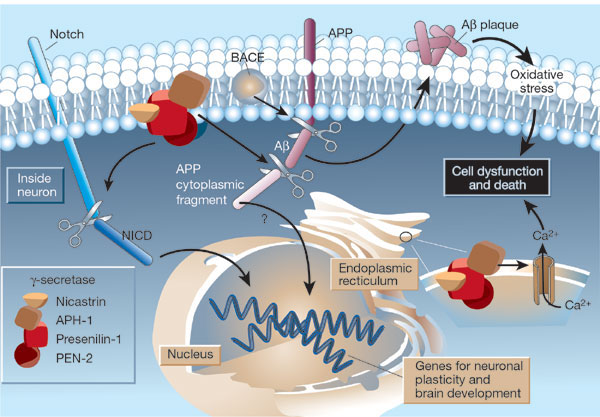
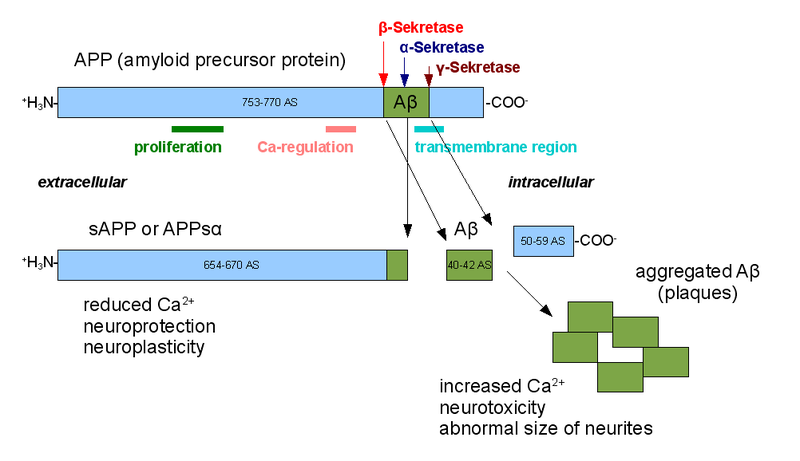 APP is cut near the cellular membrane by α-secretase enzyme. A second cut is made by γ-secretase and creates an intra cellar protein and a small extracellular peptide. In the abnormal state, a different enzyme—β-secretase—cuts APP differently, further from the membrane and leaves a fragment either 40 or 42 amino acids long. This is the famous Aβ peptide that forms clumps and then plaques. It is the 42-sized peptide that most easily forms clumps. From the familial illness, it is clear that the APP and processing are relevant to the disease. What is not clear is that even in the familial illness, that the Aβ creates the disease symptoms.
APP is cut near the cellular membrane by α-secretase enzyme. A second cut is made by γ-secretase and creates an intra cellar protein and a small extracellular peptide. In the abnormal state, a different enzyme—β-secretase—cuts APP differently, further from the membrane and leaves a fragment either 40 or 42 amino acids long. This is the famous Aβ peptide that forms clumps and then plaques. It is the 42-sized peptide that most easily forms clumps. From the familial illness, it is clear that the APP and processing are relevant to the disease. What is not clear is that even in the familial illness, that the Aβ creates the disease symptoms.
A mutation of the APP protects against AD, not a mutation in one of the secretase molecules.
APOE is a molecule that transports lipids generally, but also transports Aβ. Another curiosity is that in the sporadic form of AD, mutations in secretase or APP are relevant as they are in the genetic familial type. Why only APOE?
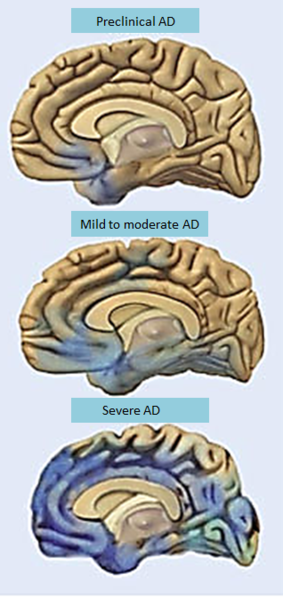 One of the problems is that there is no real way to diagnose it except with symptoms. By the time cognitive symptoms appear, it is too late. The assumption that measuring amyloid in the blood determines a symptom is not at all clear.
One of the problems is that there is no real way to diagnose it except with symptoms. By the time cognitive symptoms appear, it is too late. The assumption that measuring amyloid in the blood determines a symptom is not at all clear.
A large number of elderly people (up to a third of the population) have large amounts of Aβ in their brain without dementia. Amyloid is not sufficient to cause Alzheimer’s. Also, it took many years for this amyloid to appear in their brains. When Aβ is added to mice, Alzheimer’s doesn’t appear. There are no abnormal tau tangles and no loss of synapses. While Aβ does produce some brain toxicity, it is not enough to start Alzheimer’s. A major problem with all mouse models is that the “Alzheimer’s” that they produce is not the same as the real disease and is reversible.
Another big problem with the amyloid hypothesis is research where amyloid is eliminated by antibodies. After years of follow up, these people had much of their amyloid removed, but their cognitive problems did not improve. In fact, their dementia appears worse. This shows that there can be dementia with no plaques.
The current approach is circular and illogical. Amyloid without symptoms is defined as early Alzheimer’s without proof.
Many Different Theories Need to be Explored
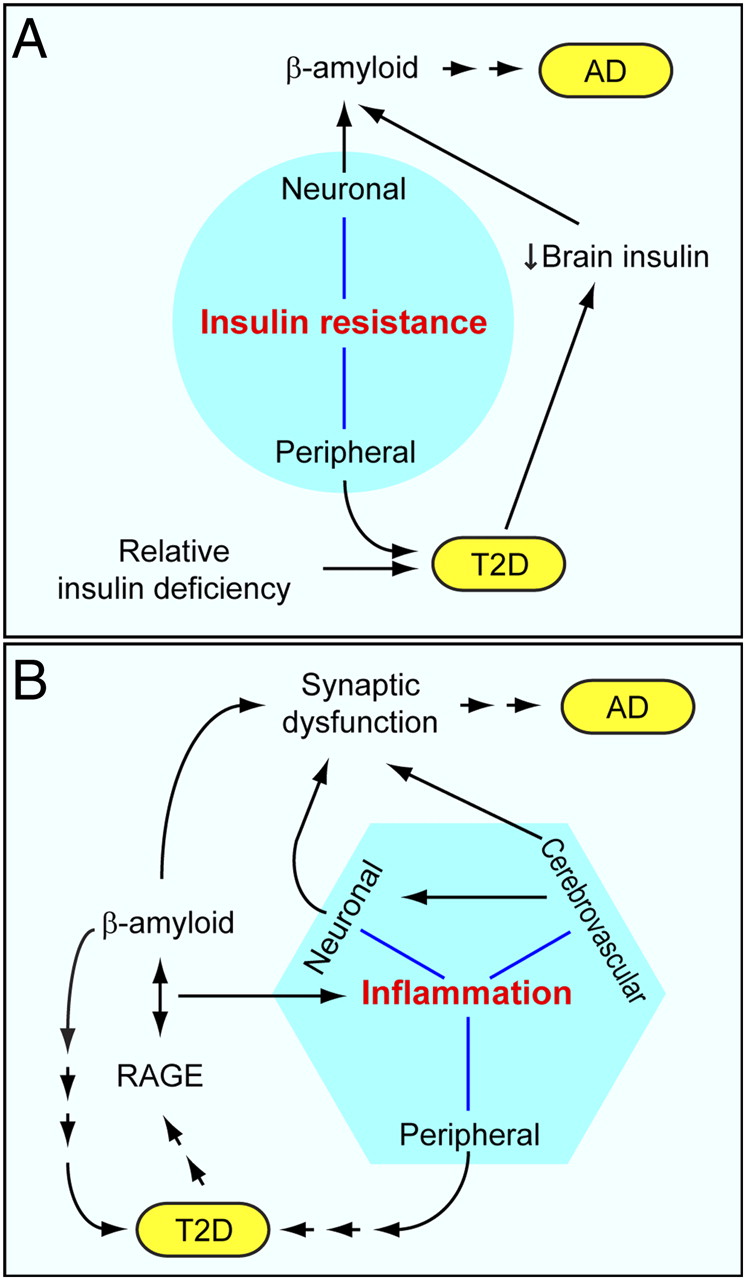 There are many factors that could be critical in the production of Alzheimer’s, all ocuring simultaneously. All of these need research, not just amyloid.
There are many factors that could be critical in the production of Alzheimer’s, all ocuring simultaneously. All of these need research, not just amyloid.
- Oxidative, DNA or mitochondrial damage
- Amyloid
- Tauopathy
- Failure of autophagy and lysosomes
- Loss of calcium homeostasis,
- Excitotoxicity from glutamate
- Failure of neuronal cell cycle
- Neuro-inflammation
Summery of Factors in Favor of the Amyloid Hypothesis
In the following AD stands for Alzheimer’s disease. A summary of factors in favor of the amyloid Hypothesis is:
 In familial, APP and PSEN genes only
In familial, APP and PSEN genes only- In sporadic, APOE mutation affects Aβ transport and AD risk
- Rare APP mutation lowers AD risk and decreases Aβ
- Amyoid related to PSEN with gamma secretase
- Longer peptide 42 favor plaque and AD
- More APP causes more plaques
- Mice have memory problems with APP mutation
- Aβ is toxic by itself in neuron cultures
- More APP in flies causes brain disease
- Aβ more common in AD
- Plaques on imaging associated with AD
- Anti plaque therapies helps some
Summary of Factors Against Amyloid Hypothesis
- In familial, no alpha secretase or BACE mutations
- Sporadic show no association with the APP, PSEN, BACE
- MAPT tau causes fronto temporal damage
- Aβ not enough in mice
- Many other deficits occur
- APP increase in humans not cause tangles or AD
- PSEN mutations have no tangles or AD
- Memory deficits in mice get rapidly better
- Tangles more closely related to AD
- People with large amount of plaque can be normal
- Antibodies clearing plaque didn’t cure dementia
- Pre clinical AD not defined
- Anti plaque therapy doesn’t help many
Does Abnormal Amyloid Cause Alzheimer’s
 As many posts have shown the brain is vastly complex. Brain diseases are, also, vastly complex. Having known the unique mutation in the gene that causes Huntington’s for 25 years has not yet produced the mechanism. It does appear that mis folded proteins are critical to all of the important brain diseases—Alzheimer’s, Parkinson’s, ALS, and Huntington’s. There is evidence that mis folded proteins are transmitted in vesicles like neurotransmitters and prions in particular neuronal circuits. These circuits produce the particular disease symptoms. Brain diseases have an inverse relationship to cancer probably because of the p53 pathway that increases destruction of cells in one direction and the production of many abnormal cells in the other.
As many posts have shown the brain is vastly complex. Brain diseases are, also, vastly complex. Having known the unique mutation in the gene that causes Huntington’s for 25 years has not yet produced the mechanism. It does appear that mis folded proteins are critical to all of the important brain diseases—Alzheimer’s, Parkinson’s, ALS, and Huntington’s. There is evidence that mis folded proteins are transmitted in vesicles like neurotransmitters and prions in particular neuronal circuits. These circuits produce the particular disease symptoms. Brain diseases have an inverse relationship to cancer probably because of the p53 pathway that increases destruction of cells in one direction and the production of many abnormal cells in the other.
Many posts have described the close connection of brain and immune systems. Clearly, inflammation either produces or follows the damage in these diseases. A particular type of microglia is produced that is involved in negative activity, while the ordinary type is critical for normal neuronal function, defense against microbe attacks and cleanup of mis folded proteins and other debris. With such complexity, treatment might need multiple approaches at once.
There has to be a wide variety of different research approaches, not just one theory.